TO: Administrative File: CAG #00445N
FROM: Tamara Syrek Jensen, JD
Director, Coverage and Analysis Group
Joseph Chin, MD, MS
Deputy Director, Coverage and Analysis Group
Lori Ashby, MA
Director
Division of Medical and Surgical Services
Kimberly Long
Lead Analyst
Jyme Schafer, MD, MPH
Lead Medical Officer
Rosemarie Hakim, PhD
Epidemiologist
SUBJECT: Proposed Decision Memorandum for Percutaneous Left Atrial Appendage Closure (LAAC) Therapy
DATE: November 10, 2015
I. Proposed Decision
A. The Centers for Medicare & Medicaid Services (CMS) proposes that the evidence is sufficient to determine percutaneous left atrial appendage closure (LAAC) therapy using an implanted device is not reasonable and necessary to diagnose or treat an illness or injury or to improve the functioning of a malformed body member and, therefore, is not covered under § 1862(a)(1)(A) of the Social Security Act.
B. In order to support evidence development for technologies likely to show benefit for the Medicare population, CMS proposes to cover items or services that are reasonable and necessary for research under §1862(a)(1)(E) of the Social Security Act using the Coverage with Evidence Development (CED) paradigm. We propose that coverage would be limited to items and services in clinical studies that meet the conditions specified below:
Percutaneous LAAC therapy is covered for patients with non-valvular atrial fibrillation only, when all of the following conditions 1-7 are met.
- The device is FDA approved for patients with non-valvular atrial fibrillation.
- The patient has:
- A high CHADS2 (Congestive heart failure, Hypertension, Age >75, Diabetes, Stroke/transient ischemia attack/thromboembolism) or CHA2DS2-VASc score (Congestive heart failure, Hypertension, Age ≥ 65, Diabetes, Stroke/transient ischemia attack/thromboembolism, Vascular disease, Sex category); and
- A high HAS-BLED score (Hypertension, Abnormal renal function and/or liver function, Stroke, prior Bleeding, Labile anticoagulation range, Elderly age>65, Drug therapy such as antiplatelet drugs), and
- A contraindication to warfarin.
- The procedure is furnished in a hospital that meets the following institutional requirements:
- Cardiac catheterization lab or electrophysiology (EP) lab with fluoroscopy capability
- Non-invasive imaging (i.e. transesophageal echocardiography) with dedicated echocardiography support
- Anesthesiology support for administration of general anesthesia specific to this procedure
- Cath lab, operating room (if required), post anesthesia recovery, intensive care and step down unit space to accommodate cases with and without complications
- On-site emergency cardiac surgery services
- The procedure is performed by physicians with the following qualifications and experience:
- The primary implanting physician has performed ≥ 25 interventional cardiac procedures involving a trans-septal puncture (TSP) in their total experience, with at least 10 TSP procedures performed over the past 12 month period.
- The primary implanting physician(s) must be an interventional cardiologist and/or an electrophysiologist. They may jointly participate in intra-procedural aspects of the implant or perform the implant procedure individually.
- The interventional cardiologist(s) and electrophysiologist(s) must receive the training prescribed by the manufacturer on the safe and effective use of the device prior to performing implant procedures. Training should also include a physician who already has experience implanting the device, but who is not a representative of the manufacturer, as well as a minimum of two supervised and two observed cases.
- The patient is enrolled in, and the treating physician team is participating in a prospective national registry that consecutively enrolls LAAC patients and tracks the following annual outcomes at the patient data level for a period of at least five years from the time of the LAAC procedure.
- Operator-specific complications
- Device-specific complications including device thromboses
- Stroke, adjudicated, by type
- TIA
- Systemic embolism
- Death
- Major bleeding, by site and severity
The registry must be designed to permit identification and analysis of patient, practitioner and facility level factors that predict patient risk for these outcomes. The registry must include contemporaneous patients followed on oral anticoagulant (OAC) therapy to serve as non-interventional controls. CMS will review the qualifications of candidate registries to ensure that the approved registry follows standard data collection practices and collects data necessary to evaluate the patient outcomes specified above.
The registry must collect all data necessary to conduct multivariable adjusted analyses and have a written executable analysis plan in place to address the following questions:
In a prospective, clinical study, when percutaneous LAAC therapy using an implanted device is performed outside a randomized, controlled clinical trial, compared to non-interventional controls followed on oral anticoagulant (OAC) therapy:
- How do the outcomes listed above compare to outcomes in the pivotal clinical trials in the short term (<12 months) and in the long term (≥ 5 years)?
- How do the outcomes listed above differ from outcomes in comparable concurrent OAC controls in the short term (<12 months) and in the long term (>5 years)?
- What is the long term (≥ 5 year) durability of the device?
- What are the short term (<12 months) and the long term (>5 years) device-specific complications including device thromboses?
- A formal shared decision-making interaction between the patient and provider using an evidence-based decision tool on anticoagulation in patients with NVAF must occur prior to LAAC, must be documented in the medical records, must include a discussion of the benefits and harms, must document an appropriate rationale to seek a non-pharmacologic alternative to anticoagulants, taking into account the safety and effectiveness of the device compared to anticoagulants, and have, after being informed of the reported risks of LAAC and reasonable alternative management strategies, given informed consent.
All CMS-approved clinical studies and registries must adhere to the following standards of scientific integrity and relevance to the Medicare population:
- The principal purpose of the study is to test whether the item or service meaningfully improves health outcomes of affected beneficiaries who are represented by the enrolled subjects.
- The rationale for the study is well supported by available scientific and medical evidence.
- The study results are not anticipated to unjustifiably duplicate existing knowledge.
- The study design is methodologically appropriate and the anticipated number of enrolled subjects is sufficient to answer the research question(s) being asked in the National Coverage Determination.
- The study is sponsored by an organization or individual capable of completing it successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found in the Code of Federal Regulations (CFR) at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it is also in compliance with 21 CFR Parts 50 and 56. In addition, to further enhance the protection of human subjects in studies conducted under CED, the study must provide and obtain meaningful informed consent from patients regarding the risks associated with the study items and/or services, and the use and eventual disposition of the collected data.
- All aspects of the study are conducted according to appropriate standards of scientific integrity.
- The study has a written protocol that clearly demonstrates adherence to the standards listed here as Medicare requirements.
- The study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Such studies may meet this requirement only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research studies and registries are registered on the www.ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject. Registries are also registered in the Agency for Healthcare Quality (AHRQ) Registry of Patient Registries (RoPR).
- The research study protocol specifies the method and timing of public release of all prespecified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 12 months of the study’s primary completion date, which is the date the final subject had final data collection for the primary endpoint, even if the trial does not achieve its primary aim. The results must include number started/completed, summary results for primary and secondary outcome measures, statistical analyses, and adverse events. Final results must be reported in a publicly accessibly manner; either in a peer-reviewed scientific journal (in print or on-line), in an on-line publicly accessible registry dedicated to the dissemination of clinical trial information such as ClinicalTrials.gov, or in journals willing to publish in abbreviated format (e.g., for studies with negative or incomplete results).
- The study protocol must explicitly discuss beneficiary subpopulations affected by the item or service under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations in the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The study protocol explicitly discusses how the results are or are not expected to be generalizable to affected beneficiary subpopulations. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions. All other indications are nationally non-covered.
The principal investigator must submit the complete study protocol, identify the relevant CMS research question(s) that will be addressed and cite the location of the detailed analysis plan for those questions in the protocol, plus provide a statement addressing how the study satisfies each of the standards of scientific integrity (a. through m. listed above), as well as the investigator’s contact information, to the address below. The information will be reviewed, and approved studies will be identified on the CMS website.
Director, Coverage and Analysis Group
Re: LAAC Therapy CED
Centers for Medicare & Medicaid Services (CMS)
7500 Security Blvd., Mail Stop S3-02-01
Baltimore, MD 21244-1850
We are requesting public comments on this proposed determination pursuant to section 1862(l) of the Social Security Act. We are specifically interested in public comments on the use of CED in this decision. After considering the public comments, we will make a final determination and issue a final decision memorandum.
II. Background
Throughout this document we use numerous acronyms, some of which are not defined as they are presented in direct quotations. Please find below a list of these acronyms and corresponding full terminology.
ACC – American College of Cardiology
ACCP – American College of Chest Physicians
AF – Atrial fibrillation
AHA – American Heart Association
ASA – American Stroke Association
CAP – Continued Access Protocol
CHADS2 - Congestive heart failure, hypertension, age >75 years, diabetes mellitus, and previous stroke/transient ischemic attack
CHA2DS2-VASc – Congestive heart failure, hypertension, age 75 years or older, diabetes mellitus, previous stroke/TIA/thromboembolism, vascular disease, age 65- 74 years, sex category (female)
FDA – United States Food and Drug Administration
HAS-BLED – Hypertension (systolic blood pressure > 160), abnormal renal function, abnormal liver function, previous stoke, prior major bleeding or predisposition, labile INR (<60% of time in therapeutic range), age > 65, drugs predisposing to bleeding (antiplatelets, nsaids), alcohol use (>8 drinks/week)
HRS – Heart Rhythm Society
ITT – Intention to treat
LAA – Left atrial appendage
NOAC – New oral anticoagulants which excludes the oral vitamin K antagonist warfarin
NVAF – Non-valvular atrial fibrillation
OAC – Oral anticoagulants
PMA – Premarketing approval
SCAI – Society for Cardiac Angiography and Interventions
SE – Systemic embolism
TEE – Transesophageal echocardiography
TIA – Transient Ischemic Attack
TTR – Time in therapeutic range
It is important to note that the terms left atrial appendage closure (LAAC) and left atrial appendage occlusion (LAAO) are used interchangeably throughout this decision memorandum.
Stroke
Stroke continues to be a leading cause of death despite an overall decline in stroke mortality, due to lower stroke incidence and improved case-fatality rates (Go et al. 2014; Lackland et al. 2013). In those who survive a stroke, functional impairment often occurs:
- A community-based estimate of disability in a subset of stroke survivors in the Framingham Heart Study cohort revealed that at 6 months post-stroke 50% had hemiparesis (weakness on one side of the body), 46.2% had cognitive deficits, 18.9% had aphasia (difficulty in expressing oneself when speaking, trouble understanding speech, and difficulty with reading and writing), 30.8% were unable to walk unassisted, 22.2% had bladder incontinence, and 29.9% had social disability (Kelly-Hayes et al. 2003).
- A large observational study from the Swedish stroke registry found activity of daily living (ADL) dependency rates at 3 and 12 months were 16.2% and 28.3% respectively (Ullberg et al. 2015).
- A systematic review of observational studies estimated 33% of all survivors experienced depression (Hackett et al. 2005).
- In a study of 11,900, visual impairment persisted in 21 % measured 90 days after stroke (Ali et al. 2013).
In review of clinical studies, it is important to understand what definition of stroke was used, i.e., case ascertainment and classification. Stroke has not been consistently defined in the past (Sacco et al. 2013). This circumstance is problematic in interpretation of clinical studies and can lead to erroneous conclusions. Sacco et al. provides an update of the current definition of stroke:

Page 2066. Table 1. Definition of stroke. Sacco et al. An updated definition of stroke for the 21st century. A statement for healthcare professionals from the American Heart Association and the American Stroke Association. Stroke 2013;44:2064-2089.
Stroke prevention activities aim to reduce risk. A number of stroke risk factors have been identified, though elevated blood pressure may be the most important determinant of the risk of stroke (Lackland et al. 2013). “It is estimated that the overwhelming majority of strokes each year could be prevented through awareness, optimal management of hypertension, and lifestyle changes to healthier diet, greater physical activity, and smoking cessation. These factors plus waist-to-hip ratio account for 82% and 90% of the population-attributable risk for ischemic stroke and hemorrhagic stroke, respectively” (Lackland et al. 2013; O’Donnell et al. 2010). To accurately reflect an individual’s stroke risk in an attempt to improve prevention through modification of treatment and/or lifestyle, stroke risk assessment tools have been developed based on modeling. However, “An ideal stroke risk assessment tool that is simple, is widely applicable and accepted, and takes into account the effects of multiple risk factors does not exist” (Meschia et al. 2014).
Atrial fibrillation as a stroke risk factor
AF is a heart arrhythmia characterized by disorganized atrial electrical activity and a loss of coordinated atrial contraction. The mechanisms and etiology of AF are incompletely understood. A number of cardiovascular diseases such as coronary heart disease, hypertension, heart failure, and valvular heart disease are antecedent to AF (January et al. 2014; Ball et al. 2013). A variety of other risk factors and biomarkers that are associated with AF including diabetes, obesity, genetic factors, smoking, and inflammation (January et al. 2014; Wyse et al. 2014; Ball et al. 2013).
Estimates on the incidence and prevalence of AF vary but it is known that AF, similar to stroke, increases with age. A Medicare claims study estimated an incidence of 28.3 per 1000 person-years and a prevalence of 85.8 per 1000 person-years for 2007 (Piccini et al. 2012). AF can be classified by duration and association with valvular mitral disease.

Page 2253. Table 3. Definitions of AF: A simplified scheme. January et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014;64:2246–80.
AF is a significant risk factor for ischemic stroke and systemic thromboembolism (SE). In AF, there is evidence to support thrombus formation from fulfillment of Virchow’s triad: (1) abnormal stasis in the left atrium causing flow abnormities; (2) structural heart and vascular disease (including endothelial or endocardial damage or dysfunction); and (3) abnormal coagulation and fibrinolysis causing a prothrombotic state (Watson et al. 2009; Lip and Lane 2015). An estimate of approximately 10% of all strokes are thought to be attributable to atrial fibrillation (Meschia et al. 2014; Wolf et al. 1991). Recent estimates from a large Swedish population-based study for attributable risk for ischemic stroke (Bjorck et al. 2013):
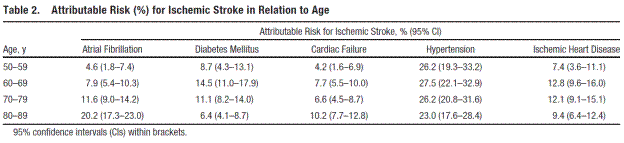
Page 3106. Table 2. Attributable Risk (%) for ischemic stroke in relation to age. Björck et al. Atrial fibrillation, stroke risk, and warfarin therapy revisited: a population-based study. Stroke. 2013;44:3103-3108.
In strokes due to NVAF, it is presumed that 90% of thromboembolisms that originate in left atria are from the left atrial appendage (LAA)based on echocardiographic and anatomic (operation and autopsy) studies that found when thrombus is observed in the left atrium in patients with AF, it is located in the LAA in >90% of cases (Holmes and Schwartz 2009; Whitlock et al. 2009; Blackshear and Odell 1996). The LAA is derived from the left wall of the atrium, which forms during the fourth week of embryonic development (Al-Saady et al. 1999). Distinct from the left atrium of the heart, it has unique developmental, anatomical, and physiological properties (Al-Saady et al. 1999). The variable structure of an individual’s LAA is generally a long, tubular, internally and externally complex structure that has a narrow junction and opening with the venous part of the left atrium. In theory this structure becomes important with age, as the incidence of AF increases with age and conditions in the LAA predispose to thromboembolism.
Stroke prevention in patients with NVAF
Within the NVAF population the risk of stroke varies widely based on the presence of other risk factors. The absolute risk is highest for those patients with a history of stroke and transient ischemic attack (TIA). Risk estimation tools have been developed to assist in treatment decisions; however the risk estimates from various tools can vary widely (Culebras 2014). Most of the schemes based on clinical criteria have weak predictive value for high-risk patients who sustain events (Lip and Lane 2015). The CHA2DS2 -VASc score has demonstrated in studies to be best at identifying patients for whom the absolute risks of stroke or systemic embolism (SE) are less than 1% per year, i.e., low risk (Lip and Lane 2015). Lip and Lane provide a comparison table of the more commonly used stratification tools:
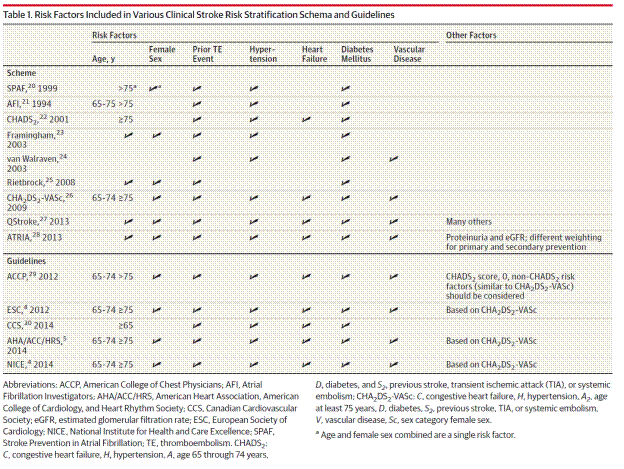
Page 1952. Table 1. Risk factors included in various clinical stroke risk stratification schema and guidelines. Lip and Lane. Stroke prevention in atrial fibrillation. A systematic review. JAMA 2015;313 (19):1950-1962.
Pharmacotherapy
Treatment with warfarin reduces the risk of stroke in NVAF by 64% (95% CI, 49-74) (Hart et al. 2007). Anticoagulation also reduces stroke severity and post stroke mortality. Anticoagulation control is directly related to efficacy, with anticoagulation control measured by time in therapeutic range (TTR). The risk of bleeding complications is closely related to therapeutic efficacy. The most serious bleeding complication is intracranial hemorrhage (ICH). Four new oral anticoagulants (dabigatran, rivaroxaban, apixaban, and edoxaban) are approved for stroke prevention in patients with NVAF. The advantages of these agents are fixed dosing, lack of required blood monitoring, fewer drug interactions, and most importantly an apparent lower risk of ICH as compared to adjusted dose warfarin. The major drawback at this time is lack of an agent to reverse their anticoagulant effects.
Preventive therapy with antiplatelet agents is less effective than anticoagulation agents. A meta-analysis of trials of antiplatelet agents (aspirin vs. placebo, aspirin vs. no treatment, aspirin plus low-dose warfarin vs. control, dipyridamole vs. placebo, dipyridamole plus aspirin vs. placebo) had a risk reduction of 22% (95% CI, 6-35) (Hart et al. 2007). In comparison to aspirin, dual antiplatelet therapy of clopidogrel and aspirin, in those deemed unsuitable for warfarin anticoagulation, resulted in a modest additional reduction but also resulted in an increase in bleeding (Connolly et al. 2009). Antiplatelet therapy appears even less effective in the elderly (vanWalraven et al. 2009).
A summary by Lip and Lane compares current guidelines from several organizations:

Page 1953. Table 2. Summary of guideline recommendations. Lip and Lane. Stroke prevention in atrial fibrillation. A systematic review. JAMA 2015;313(19):1950-1962.
Therapies directed at the LAA
Limitations with current medical therapies have led to interest in the idea of LAA exclusion to reduce the risk of thromboembolism. Three approaches for excluding the LAA include: (1) surgical approaches directed at amputation or ligation of the LAA; (2) percutaneous endovascular strategies that place a device to attempt occlusion of the structure; and (3) percutaneous epicardial ligation aimed at external LAA occlusion (Sakellaridis et al. 2014).
The procedure to implant an occluding device percutaneously has been similar in the devices to date. After venous access, the septum is punctured for access to the left atrium. The implant is advanced under transesophageal echocardiography (TEE) and/or intracardiac echocardiography, fluoroscopic or angiographic guidance and is deployed into the LAA opening to effectively occlude the opening. The angulation, length, and number of lobes of the LAA as well as opening size can vary necessitating estimation of proper device size as the LAA has a variable size and morphology. While several devices have been developed, only one device is currently FDA approved.
Percutaneous epicardial occlusion involves ligating the LAA at its ostium. Currently no device has received FDA approval for this use. Open surgical approaches are utilized for patients undergoing mitral valve surgery or as an adjunct to the maze procedure.
III. History of Medicare Coverage
CMS does not currently have an NCD on LAAC therapy.
A. Current Request
On May 21, 2015, CMS accepted a formal complete request from Boston Scientific Corporation to initiate a national coverage analysis (NCA) for percutaneous, transcatheter, intraluminal LAAC using an implanted device.
CMS opened this NCA to thoroughly review the evidence to determine whether or not percutaneous LAAC therapy in non-valvular AF may be covered nationally under the Medicare program. The scope of this NCA does not include surgical techniques used to achieve LAA closure.
B. Benefit Category
For an item or service to be covered by the Medicare program, it must fall within one of the statutorily defined benefit categories outlined in the Social Security Act. Percutaneous LAAC therapy falls under the benefit categories set forth in section §1861(b) (inpatient hospital services), a part A benefit under §1812(a)(1), and §1861(s)(1) (physician services), a part B benefit. This may not be an exhaustive list of all applicable Medicare benefit categories for this item or service.
IV. Timeline of Recent Activities
| Date |
Action |
May 21, 2015 |
CMS accepts a formal request from Boston Scientific Corporation for Percutaneous Left Atrial Appendage (LAAC) Therapy. A tracking sheet was posted on the web site and the initial 30 day public comment period commenced. |
| June 6, 2015 |
SentreHeart, Inc. met with CMS to discuss the LARIAT® procedure. |
June 20, 2015 |
The initial 30 day public comment period ended. 89 comments were received. (90 comments were received but one was submitted in duplicate) |
| July 29, 2015 |
Boston Scientific Corporation met with CMS to discuss their NCD request. |
| November 10, 2015 |
Proposed decision posted. 30 day comment period commenced. |
V. FDA Status
On March 13, 2015, the FDA approved the WATCHMAN™ LAA Closure Technology. This device is indicated to reduce the risk of thromboembolism from the LAA in patients with non-valvular atrial fibrillation who:
- Are at increased risk for stroke and systemic embolism based on CHADS2 or CHA2DS2-VASc scores and are recommended for anticoagulation therapy;
- Are deemed by their physicians to be suitable for warfarin; and
- Have an appropriate rationale to seek a non-pharmacologic alternative to warfarin, taking into account the safety and effectiveness of the device compared to warfarin.
The complete FDA approval and labeling can be accessed at
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P130013&source=govdelivery&utm_medium=email&utm_source=govdelivery.
VI. General Methodological Principles
When making national coverage determinations, CMS generally evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. The critical appraisal of the evidence enables us to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for beneficiaries. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary.
A detailed account of the methodological principles of study design that the Agency utilizes to assess the relevant literature on a therapeutic or diagnostic item or service for specific conditions can be found in Appendix A.
Public commenters sometimes cite the published clinical evidence and provide CMS with useful information. Public comments that provide information based on unpublished evidence, such as the results of individual practitioners or patients, are less rigorous and, therefore, less useful for making a coverage determination. CMS uses the initial comment period to inform its proposed decision. CMS responds in detail to the public comments that were received in response to the proposed decision when it issues the final decision memorandum.
VII. Evidence
A. Introduction
This presentation of evidence focuses upon whether current percutaneous implanted device randomized controlled trials (RCTs) are adequate to draw conclusions about health outcomes for prevention of stroke and thromboembolism, as well as whether the body of evidence is generalizable to the Medicare population. The evidence CMS examines has as its focus health outcomes, i.e., the benefits and harms of a particular treatment. Key outcomes of interest to CMS are mortality, ischemic and hemorrhagic stroke, system thromboembolism, major bleeding, and procedural and device related complications. We summarize the evidence relating to the prevention of thromboembolic events in patients with NVAF with percutaneous device therapy. In prevention of thromboembolic events in NVAF, the primary focus is risk reduction.
B. Literature Search
CMS searched PubMed for articles containing the term left atrial appendage and atrial fibrillation. The search was performed from February to June 2015. Subsequent searches include guidelines and systematic reviews on the topic of stroke prevention in patients with NVAF. Public access information from the FDA website was also used. Transcatheter placed devices with randomized trials and consecutive case series with more than 50 patients and with a minimum of 6 months follow-up reporting thromboembolic event outcomes were included, the latter included as some technologies do not have randomized trials. Studies must have been published in peer-reviewed English language journals. The literature search was limited to the English language and specific to the human population, but included studies conducted in all countries.
C. Discussion of Evidence
The development of an assessment in support of Medicare coverage decisions is based on the same general question for almost all national coverage analyses (NCAs): "Is the evidence sufficient to conclude that the application of the item or service under study will improve health outcomes for Medicare patients?" For this NCD, the questions of interest are:
Is the evidence sufficient to conclude that percutaneous left atrial appendage closure (LAAC) improves health outcomes for Medicare beneficiaries with non-valvular atrial fibrillation?
If the answer to the question above is positive, is the available evidence sufficient to identify the characteristics of the patient, practitioner or facility that predict which beneficiaries are more likely to experience overall benefit or harm from LAAC?
1. External Technology Assessment (TA)
CMS did not commission an external TA for this NCA.
Blue Cross Blue Shield Association. Percutaneous left atrial appendage closure therapy for the prevention of stroke. Technol Eval Cent Assess Program Exec Summ. 2014 Oct;29(5):1-4.
Summary of application of the technology evaluation criteria
“Based on the available evidence, the Blue Cross and Blue Shield Association Medical Advisory Panel made the following judgments about whether percutaneous left atrial appendage closure (LAAC) therapy for stroke prevention meets the Blue Cross and Blue Shield Association Technology Evaluation Center (TEC) criteria.
1. The technology must have final approval from the appropriate governmental regulatory bodies.
Currently, no percutaneous LAAC device has FDA approval. The WATCHMAN™ LAA Closure Device (Boston Scientific, Maple Grove, MN) was considered by FDA for approval in 2013 based on the results of a randomized trial (PREVAIL [Prospective Randomized Evaluation of the WATCHMAN LAA Closure Device in Patients With Atrial Fibrillation Versus Long Term Warfarin Therapy]) comparing LAAC with warfarin for prevention of stroke and thromboembolism in patients with Non-valvular atrial fibrillation (AF). At the time of this Assessment, FDA approval is pending. The Amplatzer™ Cardiac Plug (St. Jude Medical, Minneapolis, MN) has not yet been approved by FDA. The Lariat® Loop Applicator device (SentreHEART Inc., Redwood City, CA) is a suture delivery system that received FDA 510(k) marketing clearance in 2006. It is not specifically approved for LAAC.
2. The scientific evidence must permit conclusions concerning the effect of the technology on health outcomes. The evidence is insufficient to permit conclusions about the effect of the percutaneous LAAC technologies on health outcomes. For the clinical indication of stroke prevention in patients with Non-valvular AF without contraindications to anticoagulation, there are 2 randomized controlled trials (RCTs) and 1 case series. One RCT, meeting noninferiority criteria for a principal composite outcome of stroke, death, and embolism, comparing percutaneous LAAC with anticoagulation had problems with patient selection, potential confounding with other treatments, and losses to follow-up. The second RCT, which incorporated the first trial’s results as a Bayesian prior, did not meet noninferiority criteria for its first principal composite outcome (stroke, death, and embolism). A second principal outcome (stroke and embolism after 7 days) met noninferiority criteria in 1 of 2 analyses and a performance goal for short-term complication rate. However, when considering the results of both trials, it is unclear whether LAAC is noninferior to anticoagulation. The evidence provided by an additional case series, studying patients without contraindications to anticoagulation, and 4 case series, studying patients with contraindications to anticoagulation, is insufficient to permit conclusions about efficacy. Although rates of stroke and other events were lower than expected based on measured patient characteristics, potential selection biases make conclusions difficult. The case series were mostly small studies with low statistical power.
3. The technology must improve the net health outcome. The evidence is insufficient to make conclusions about improvement in net health outcomes compared with established alternatives.
4. The technology must be as beneficial as any established alternatives. The evidence is insufficient to make conclusions about improvement in net health benefit compared with established alternatives.
5. The improvement must be attainable outside the investigational settings. It has not yet been demonstrated whether percutaneous LAAC therapy improves health outcomes in the investigational setting. Therefore, it cannot be demonstrated whether improvement is attainable in a clinical setting.
Based on the above, percutaneous LAAC therapy does not meet the Blue Cross and Blue Shield Association Technology Evaluation Center (TEC) criteria.”
2. Internal technology assessment
WATCHMAN device
Background
When a trial has multiple timeframe publications, the report that analyzes the longest, most complete follow-up is in general summarized. In the instance of the WATCHMAN device, due to the adaptive trial design, all publications from various follow-up timeframes that report on the study population are herein included, unless it is a duplicative report or did not meet search inclusion criteria. The randomized trial reports are as follows: Holmes et al. August 2009 reports on the PROTECT AF fourth planned interim analysis at 1065 patient-years; Reddy et al. January 2011 reports on a PROTECT AF post-hoc analysis done to examine the effect of experience on safety and includes 1500 patient-years of follow-up and continued access registry data from 460 patients at 26 centers; Reddy et al. February 2013 reports on the PROTECT AF final planned interim analysis at 1588 patient-years; Reddy et al. 2014 reports on PROTECT AF data at 2621 patient years at a mean (SD) follow-up of 3.8 (1.7) years; Holmes et al. 2014 reports on PREVAIL data with a mean follow-up of 11.8 months, with PROTECT AF data as an informative prior. In addition, summaries of the three FDA Executive Summaries are included.
Study Design
Fountain RB, Holmes DR, Chandrasekaran K, Packer D, Asirvatham S, Tassel RV, and Turi Z. The PROTECT AF (WATCHMAN left atrial appendage system for embolic PROTECTion in patients with atrial fibrillation) trial. Am Heart J 2006;151:956-61.
The objective of sponsor funded PROTECT AF was to demonstrate safety and effectiveness of the WATCHMAN implant as compared to warfarin therapy alone in patients with NVAF who can be treated with warfarin. The effectiveness end point was defined as all stroke (ischemic and hemorrhagic), cardiovascular death (any cardiovascular death or unexplained death), systemic embolism (SE), and transient ischemia attack (TIA) (defined as an acute focal neurological event lasting at least 5 minutes). Patient selection criteria:

Page 957. Table 1. Fountain et al. The PROTECT AF (WATCHMAN left atrial appendage system for embolic PROTECTion in patients with atrial fibrillation) trial. American Heart Journal 2009; 151(5): 957-961.
The randomization scheme was 2:1 (two implants to one control) and stratified by center. Randomization was performed electronically. The statistical objective was designed for noninferiority of event rate defined as the number of expected events per 100 pt-yrs of follow-up. Patient years were to be calculated from the date of randomization for each patient and aggregated over groups. The boundaries for the noninferiority were two times the control. A Bayesian Hierarchical model is used for evaluation. The number of patients to be enrolled was determined to be a minimum of 600 patient years and a maximum of 1500 patient years to demonstrate noninferiority. The adaptive design planned for the first evaluation after 600 patient years of follow-up with subsequent evaluations after each additional 150 patient years up to a maximum of 1500 patient years. Other criteria included that at least 300 patients would be followed for 1 year and at least 100 patients would be followed for 2 years. It was planned for an ITT analysis so patients were analyzed as being in their randomized group regardless of actual treatment. Censoring at time of last follow-up occurred if patients were lost to follow-up or if patients had an event. It was planned that analyses for the effects of covariates would be performed to look at the effect baseline and procedural variables have on the study end points. Specific covariates of interest for the primary composite endpoint included CHADS2 scores, age, sex, AF category, left atrium size, and left ventricular ejection fraction. Sensitivity analyses were planned to include at a minimum a per-protocol analysis. A primary safety endpoint was defined as “Life-threatening events as determined by the Clinical Events Committee which would include events such as device embolization requiring retrieval and bleeding events e.g., pericardial effusion requiring drainage, cranial bleeding events due to any source, gastrointestinal bleeds requiring transfusion, and any bleeding related to the device or procedure that necessitates an operation.” Several secondary endpoints were defined, including technical and procedural success for the WATCHMAN group and non-therapeutic INR for the control.
Additional protocol details are included in Fountain 2009. The control INR aim was between 2 and 3, monitored on a monthly basis, “or more frequently if needed.” The implant procedure is performed under local or general anesthesia. Warfarin dose is adjusted so INR is < 2.0. Heparin is administered during the procedure. During the procedure, the specific anatomy of the LAA is evaluated including the shape of the neck, the length of the appendage, and the presence or absence of multiple lobes, and the relationship to the pulmonary veins. The size of the device is selected and then positioned and deployed. The device is imaged for positioning and absence of flow around the device into and out of the appendage. Maneuvers ensure the device is stable. An independent core laboratory was utilized to interpret all echocardiographic data. Warfarin is restarted and aspirin is continued. Patients continue on warfarin for at least 45 days. INRs are drawn weekly. At the 45 day visit TEE is repeated to assess the device position, flow through and/or around the WATCHMAN implant, stability of device position over time, atrial septal shunt from the procedure puncture, and any thrombus on the device surface or in the LAA. Warfarin is discontinued at 45 days if the TEE documents LAA occlusion (defined as no flow through the device and a residual jet flow of ≤3 mm around the margins of the WATCHMAN implant). If warfarin is discontinued, clopidogrel 75 daily is prescribed until 6 month follow-up, with aspirin 325 prescribed for the duration of the trial. Warfarin is continued in the WATCHMAN patients if TEE does not document occlusion. All patients receive assessments at 45 days, 6 months, 9 months, 12 months, and then semi-annually for the duration of the study. Device patients also have a diagnostic TEE exam at 6 and 12 month follow-up. Additional TEE exams are at the discretion of the investigators. Control and implant patients are evaluated by a neurologist at the 6 and 12 month and at the annual clinical follow-ups thereafter for 5 years. A neurological consult is performed if any clinical event occurs or if patients meet certain criteria (≥ 2 point increase on the NIHSS; ≥15 point decrease on the BI; ≥ 1 point increase on the MRS from the previous assessment, and if the change is not attributed to a nonneurological cause). Patients in both arms complete a SF12v2 Health Survey at the 12 month follow-up visit.
FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
The description of the trial design is consistent with Fountain 2006. For endpoint evaluation, a formal hypothesis was established only for the primary effectiveness endpoint, with a Bayesian model. A non-informative prior distribution was assumed. The first interim analysis at 600 patient years included 282 patients (185:97 for device: control) with at least one year of follow-up and 91 patients (60:31 for device: control) had 2 years. The reviewed data set was based on 600 patient-years, with additional data from a supplemental 900 patient-year follow-up cohort. For the 900 patient-year data, 800 patients were enrolled from 59 centers (55 U.S. and 4 European). This total includes 93 patients termed as “roll-in.” Patient accountability is summarized in the following figure:
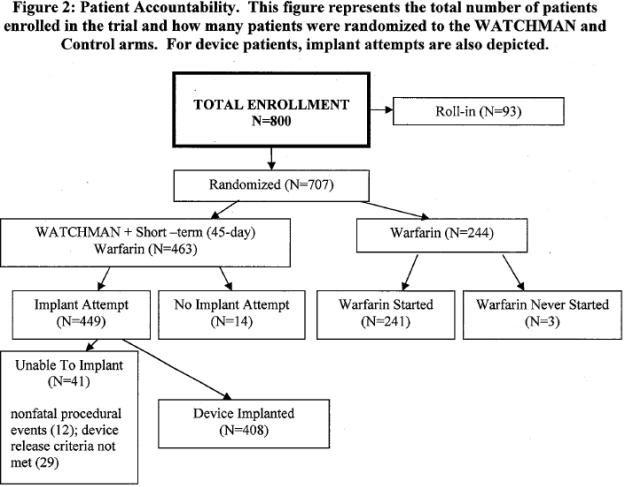
Page 16. Figure 2: Patient Accountability. This figure represents the total number of patients enrolled in the trial and how many patients were randomized to the WATCHMAN and control arms. For device patients, implant attempts are also depicted. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
Patient assessments are summarized in the following table:
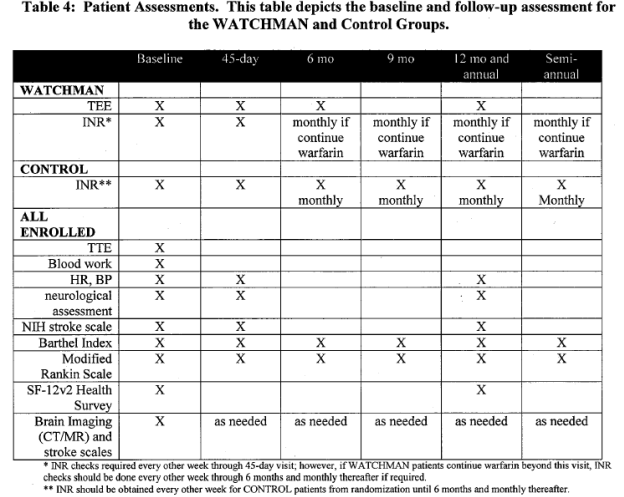
Page 13. Table 4: Patient Assessments. This table depicts the baseline and follow-up assessment for the WATCHMAN and control groups. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
Annual office visits and semi-annual telephone follow-ups were required for all patients up to 5 years unless the study was terminated or sufficient information for regulatory authorities was obtained.
Patient demographics:
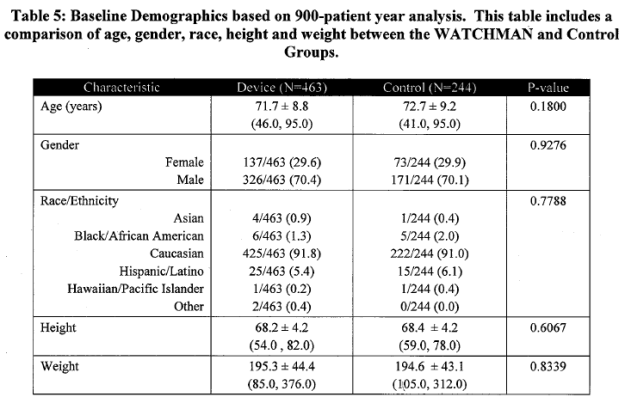
Page 17. Table 5: Baseline demographics based on 900-patient year analysis. This table includes a comparison of age, gender, race, height and weight between the WATCHMAN and control groups. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
Patient Risk Factors:
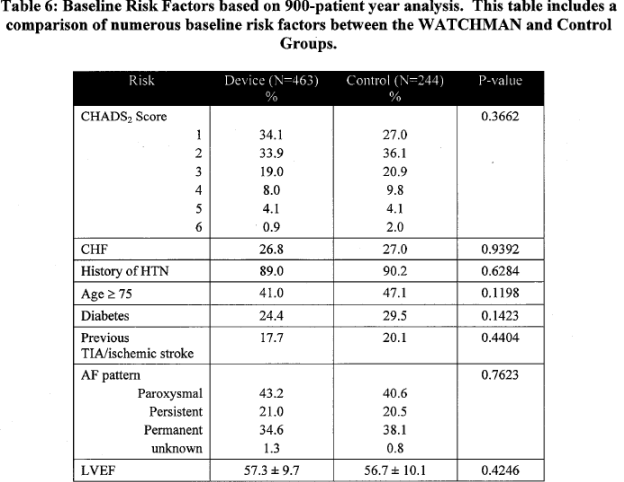
Page 18. Table 6: Baseline risk factors based on 900-patient year analysis. This table includes a comparison of numerous baseline risk factors between the WATCHMAN and control groups. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
Primary effectiveness results of the composite endpoint were analyzed for the ITT population, defined as randomization arm, regardless of treatment actually received. Composite endpoint rates are per 100 patient-years of follow-up.
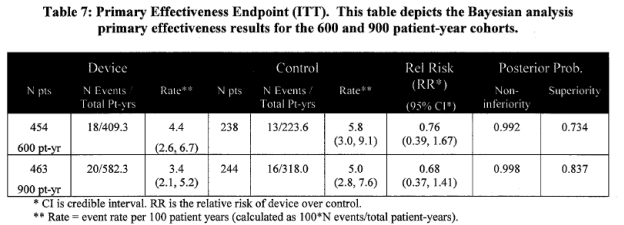
Page 19. Table 7: Primary Effectiveness Endpoint(ITT). This table depicts the Bayesian analysis primary effectiveness results for the 600 and 900 patient-year cohorts. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
The sponsor concluded that the primary effectiveness endpoint was met as the event rate in the device arm is less than two times the control rate. The upper bounds of the 95% credible interval (CI) was less than the noninferiority margin of 2.0 (1.41 for 900 pt-yr).
Accounting for the 20 events in the device arm at 900 pt-yr:
- Eleven events occurred in patients who discontinued warfarin at 45 days, with 2 of 11 restarting warfarin at a later date
- Three events occurred in patients who discontinued warfarin at a point greater than 45 days
- The remaining 6 events occurred in patients who did not receive a device, or for whom no warfarin information was available
Accounting for the 16 events in the control arm at 900 pt-yr:
- Ten occurred in patients on warfarin
- Five occurred in patients who discontinued or interrupted therapy
- One occurred in a patient without information on warfarin status
The Kaplan-Meier estimates for freedom from primary effectiveness endpoint events is found in Table 8 in the FDA document.
Individual components of the primary effectiveness composite endpoint were analyzed.
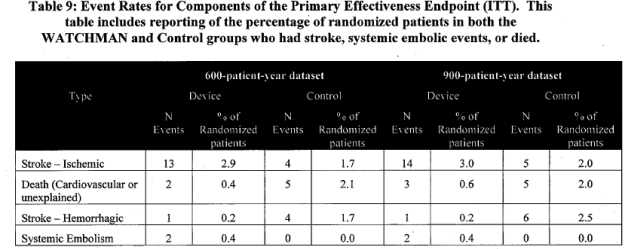
Page 21. Table 9. Event rates for components of the primary effectiveness endpoint (ITT). This table includes reporting of the percentage of randomized patients in both the WATCHMAN and control groups who had stroke, systemic embolic events, or died. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
Total trial mortality included 17 deaths in the WATCHMAN group and 15 deaths in the control. Some of the deaths were counted toward the composite endpoint, FDA table 9. For interpretation of mortality data, the FDA added these observations:


A constant primary endpoint event rate over every follow-up interval was assumed in the proposed analytic model; however non-homogeneity in event rates occurred. A Bayesian piecewise proportional hazards model with CHADS2 adjustment and a frequentist approach were used to analyze the primary effectiveness endpoint data. While it is noted that difficulties with the non-homogeneous rate can’t be overcome, both analyses were supportive of the two-fold noninferiority assumption.
A per-protocol analysis was prespecified by the sponsor. The FDA stated, “This analysis biases the results towards the device group because of the exclusion of a large proportion the device adverse events and the introduction of selection bias.” This “post-procedure” analysis differentially excluded any patient with an adverse event that occurred either before or on the date of device implant. In regards to this analysis, “The FDA does not view this analysis as clinically meaningful.”
Warfarin therapy in both arms:

Page 26. Table 13: Warfarin discontinuation from date of implantation – device arm (FDA’s calculation). This table depicts reasons for discontinuation of warfarin in the device arm and the number of patients in each category. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.

Page 26. Table 14: Warfarin discontinuation – control arm (FDA’s calculation). This table depicts reasons for discontinuation of warfarin in the control arm and the number of patients in each category. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
Calculations to examine the warfarin discontinuation impact showed a non-statistical increase in events in the group that discontinued warfarin in the 600 patient-year sample and a slightly higher event rate in the group who continued warfarin in the 900 patient-year sample. Results may not be reliable due to small sample size, with between group differences possibly due to differential follow-up, and differences in event rates per follow-up interval.
The impact of warfarin therapy in the incidence of ischemic stroke and systemic embolism was evaluated:
![Page 28. Table 16: Device ischemic strokes and systemic emboli categorized by procedure-related and warfarin status. This table[s] depicts the number of pre-procedure events, events temporally related to the procedure, and events not temporally related to the procedure, including which events occurred on and off warfarin. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.](http://www.cms.gov/medicare/coverage/determinationprocess/other-content-types/id281image036gif.gif)
Page 28. Table 16: Device ischemic strokes and systemic emboli categorized by procedure-related and warfarin status. This table[s] depicts the number of pre-procedure events, events temporally related to the procedure, and events not temporally related to the procedure, including which events occurred on and off warfarin. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
It is noted that device patients remained on warfarin 19% (600 pt-yrs) and 23% (900 pt-yrs) of follow-up time. In both arms, patients taking warfarin had therapeutic INRs only about half of the time.
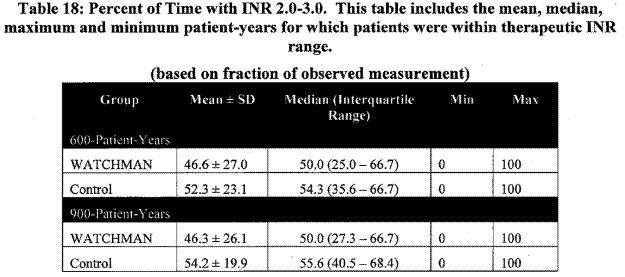
Page 29. Table 18: Percent of time with INR 2.0 – 3.0. This table includes the mean, median, maximum and minimum patient-years for which patients were within therapeutic INR range. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
Patients in the device arm were to be on clopidogrel through six months if they discontinued warfarin, and aspirin through the duration of the study. Other than the previous protocol requirements, antiplatelet therapy was at the discretion of the investigator. The percentage of follow-up time that patients were on clopidogrel was higher in the device group compared to the control group (51% versus 16%) and similarly with aspirin (91% versus 54%, device group versus control). Data were not available on the number of events on one or two platelet medication; however some control patients who had endpoint events were on warfarin and at least one antiplatelet medication.
The primary safety endpoint did not have a hypothesis. It included peri-procedural events and certain bleeding events. Some events were counted for the efficacy endpoint only, some were counted for the safety endpoint only and some were counted for both. The majority of device events, 56%, occurred on the day of the procedure. Safety events in the control group occurred later and on a continuing basis. Of the six hemorrhagic strokes in the control group (captured as effectiveness events) 4 resulted in death. For 2 of those 6 patients in the control group, warfarin status was unknown.
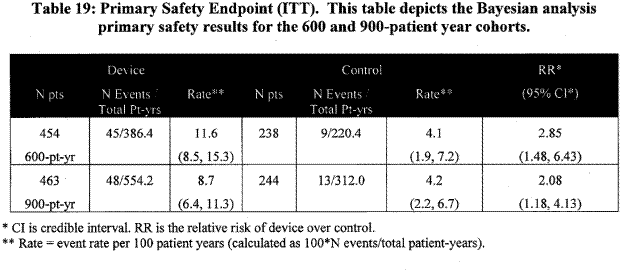
Page 30. Table 19: Primary safety endpoint (ITT). This table depicts the Bayesian analysis primary safety results for the 600 and 900-patient year cohorts. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
The Kaplan-Meier estimates for freedom from primary safety endpoint events for the ITT population is in FDA Table 20, showing that most of the events for the device group occurred within 7 days of the procedure whereas the warfarin events in general occurred later in the trial. Safety event by type:
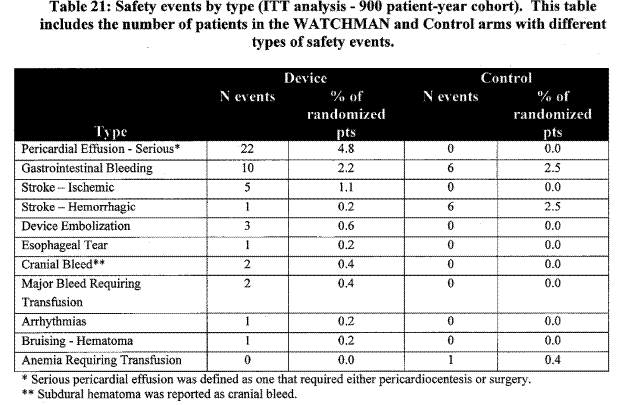
Page 32. Table 21: Safety events by type (ITT analysis – 900 patient-year cohort). This table includes the number of patients in the WATCHMAN and the control arms with different types of safety events. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
The FDA analyzed how the delay between randomization and implantation affected the data, as the time to event was based on randomization date. The relative risks reported by the applicant increased slightly but not significantly. Another analysis included this effect of only counting a patient’s first event:
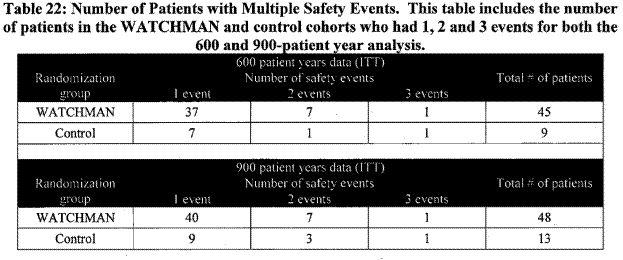
Page 33. Table 22: Number of patients with multiple safety events. This table includes the number of patients in the WATCHMAN and control cohorts who had 1,2, and 3 events for both the 600 and 900-patient year analysis. FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
The FDA calculated primary safety event rates for the device arm by warfarin discontinuation as the 45 day visit:
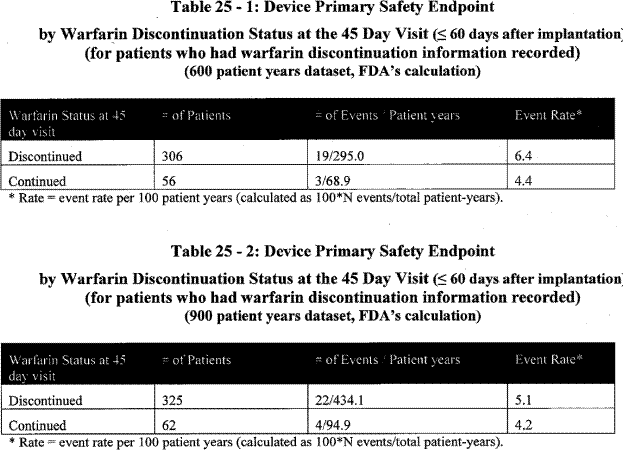
Page 34. Table 25-1: Device primary safety endpoint by warfarin discontinuation status at the 45 day visit (≤ 60 days after implantation (for patients who had warfarin discontinuation information recorded) (600 patient years dataset, FDA’s calculation)
Table 25-2: Device primary safety endpoint by warfarin discontinuation status at the 45 day visit (≤ 60 days after implantation (for patients who had warfarin discontinuation information recorded) (600 patient years dataset, FDA’s calculation). FDA Executive Summary Memorandum. Prepared for the April 23, 2009 meeting of the Circulatory System Devices Advisory Panel. P080022. WATCHMAN LAA Closure Technology, Atritech, Inc.
There were a number of additional safety considerations during this trial. In the 900 patient-year cohort, 42.6% of patients had an INR ≥ 4 at least once during follow-up. Thrombus was identified on the device in 15 patients, one of whom had a cerebral ischemia endpoint. The true incidence of subclinical cerebral infarcts is not known. Device embolization occurred in 3 patients, 2 of whom were asymptomatic. The device was explanted in two patients. Associated morbidity was significant. Only 58% of devices were adequately placed on the first attempt, 42% requiring one or more recaptures with approximately 4% recaptured four or more times. Device malfunctions occurred in 23 patients. An analysis demonstrated a learning curve with incidence of pericardial effusions and device recaptures.
In summary, the FDA noted a number of confounding issues regarding the interpretation of the favorable statistical conclusion of the primary statistical analysis of effectiveness.
Fourth planned interim analysis. 1065 patient years.
Holmes DR, Reddy VK, Turi ZG, Doshi SK, Sievert H, Buchbinder M, Mullin CM, Sick P, for the PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomized non-inferiority trial. Lancet 2009;374:534-42.
Holmes et al. reports the pre-specified fourth interim analysis at 1065 patient years. Randomization is as previously described and was done with a centralized system with block sizes of six (4 device: 2 control). An independent statistician generated the randomization sequence. The centralized computer system was password protected and accessed by the principal investigator or study coordinator after the patient gave consent and had met inclusion criteria. Participants and clinicians were not masked to treatment assignment. Participants, methods, and pre-specified analyses are as mentioned in prior summaries.
Trial enrollment:
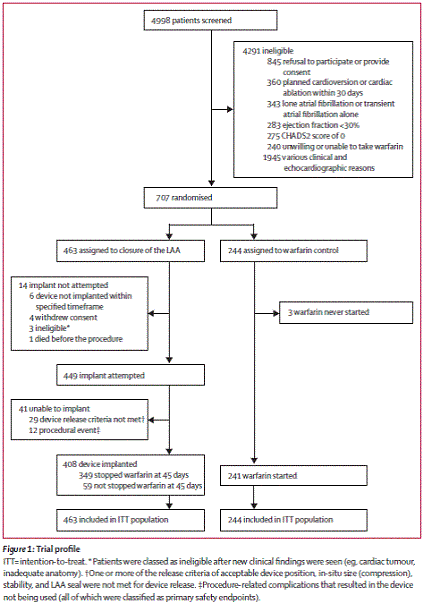
Page 535. Figure 1: Trial profile Holmes DR et al. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomized non-inferiority trial. Lancet 2009;374:534-42.
Clinical outcomes are summarized below:
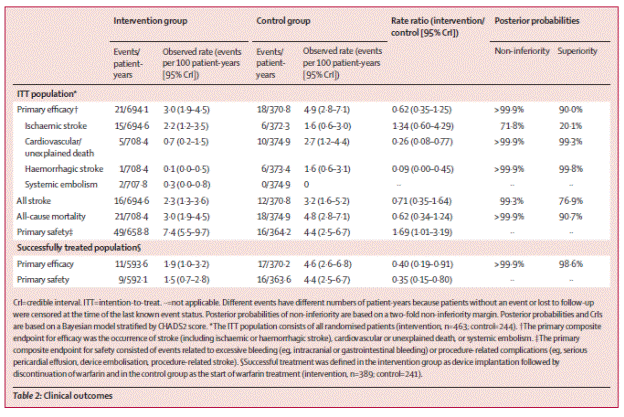
Page 537. Table 2. Clinical outcomes. Holmes DR et al. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomized non-inferiority trial. Lancet 2009;374:534-42.
The authors summarize, “Thus, our strategy for closing the LAA was non-inferior to warfarin therapy in terms of the primary efficacy endpoint of all stroke, cardiovascular death, and systemic embolism. Although there is a higher initial safety event rate for device implantation, adverse events were without long-term sequelae for most patients. Closure of the LAA might provide an alternative strategy to chronic warfarin therapy for stroke prophylaxis in patients with non-valvular atrial fibrillation.”
Post-hoc analysis with data on patients from PROTECT AF trial followed up for a total of 1500 patient-years and data from patients in the Continued Access Protocol (CAP) Registry from 460 patients
Reddy VY, Holmes D, Doshi SK, Neuzil P, and Kar S. Safety of percutaneous left atrial appendage closure. Results from the WATCHMAN left atrial appendage system for embolic protection in patients with AF (PROTECT AF) clinical trial and the continued access registry. Circulation 2011;123:417-424.
The investigators report the influence of experience on the safety of percutaneous LAA closure in a post-hoc analysis. This analysis included data from 542 patients in PROTECT AF for a total of 1500 patient-years and 460 patients in the CAP Registry. The CAP registry was designed to allow continued access to the WATCHMAN device. The registry had the same inclusion and exclusion criteria and procedure/treatment protocol as PROTECT AF. Registry patients included patients enrolled between August 2008 and April 2010 from 26 centers. The registry data does not include all the centers that participated in the PROTECT AF trial but 26 of the higher-enrolling centers. Baseline patient demographics reveal differences in mean age and CHADS2 scores.
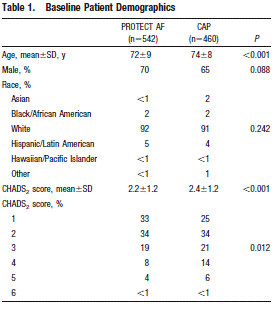
Page 419. Table 1. Baseline patient demographics. Reddy VY et al. Safety of percutaneous left atrial appendage closure. Results from the WATCHMAN left atrial appendage system for embolic protection in patients with AF (PROTECT AF) clinical trial and the continued access registry. Circulation 2011;123:417-424.
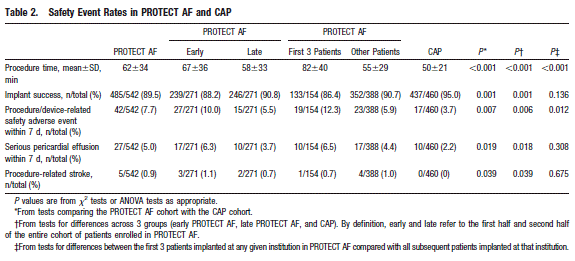
Page 419. Table 2. Safety event rates in PROTECT AF and CAP. Reddy VY et al. Safety of percutaneous left atrial appendage closure. Results from the WATCHMAN left atrial appendage system for embolic protection in patients with AF (PROTECT AF) clinical trial and the continued access registry. Circulation 2011;123:417-424.
The authors conclude, “As with all interventional procedures, there is a significant improvement in the safety of WATCHMAN left atrial appendage closure with increased operator experience.”
Final planned interim analysis at 1588 patient-years
Reddy VK, Doshi SK, Sievert H, Buchbinder M, Neuzil P, Huber K, Halperin JL, Holmes D, on behalf of the PROTECT AF Investigators. Percutaneous left atrial appendage closure for stroke prophylaxis in patients with atrial fibrillation. 2.3 year follow-up of the PROTECT AF (WATCHMAN left atrial appendage system for embolic protection in patients with atrial fibrillation) trial. Circulation 2013; 127:720-729.
The authors report on the final planned interim analysis at 1588 pt-yrs (1025.7 in the device group and 562.7 in the control group). They note that trial sample size was estimated on the basis of data from the Stroke Prevention in Atrial Fibrillation study, with an expected primary efficacy event rate of 6.15% per year in the warfarin group. Simulations were performed to ensure 80% power and 5% type I error rate in this Bayesian adaptive trial design. A one-sided noninferiority probability criterion of at least 97.5% was selected with the use of a 2-fold noninferiority margin based on the ratio of primary efficacy event rates. The mean follow-up interval for this report was 2.3 ± 1.1 (range 0-5.9 years) and median follow-up was 2.4 years with 469 patients completing 2 years of follow-up (319 in the device group and 150 in the control group). Study discontinuation rates were 15.3 % (71/463) in the device group and 22.5% (55/244) in the warfarin group. Time in therapeutic range for the warfarin group was reported as 66%.
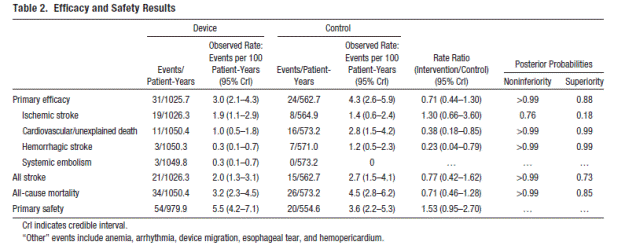
Page 723. Table 2. Efficacy and safety results. Reddy VK et al. Percutaneous left atrial appendage closure for stroke prophylaxis in patients with atrial fibrillation. 2.3 year follow-up of the PROTECT AF (WATCHMAN left atrial appendage system for embolic protection in patients with atrial fibrillation) trial. Circulation 2013; 127:720-729.
The authors conclude, “This final analysis of the entire PROTECT AF trial cohort followed up for an accumulated exposure of 1588 patient-years revealed closure of the LAA with the WATCHMAN device to be noninferior to ongoing warfarin therapy with regard to prevention of stroke, systemic embolism, and cardiovascular death.”
Continued follow-up at a mean (SD) of 3.8 (1.7) years, or 2621 patient-years.
ReddyVK, Sievert H, Halperin J, Doshi SK, Buchbinder M, Neuzil P, Huber K, Whisenant B, Kar S, Swarup V, Gordon N, Holmes D, for the PROTECT AF Steering Committee and Investigators. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation. A randomized clinical trial. JAMA 2014;312(19):1988-1998.
The authors report on a mean (SD) follow-up of 3.8 (1.7) years (2621 pt-yrs). Study design and methods are as previously described. Additional information is as follows: Patients were enrolled from February 2005 to June 2008. Potential study candidates were identified in both the outpatient and inpatient setting by physician or self-referrals in response to approved study announcements. The study cohort included 59 sites in the U.S. and Europe; one site was censored due to data quality concerns.
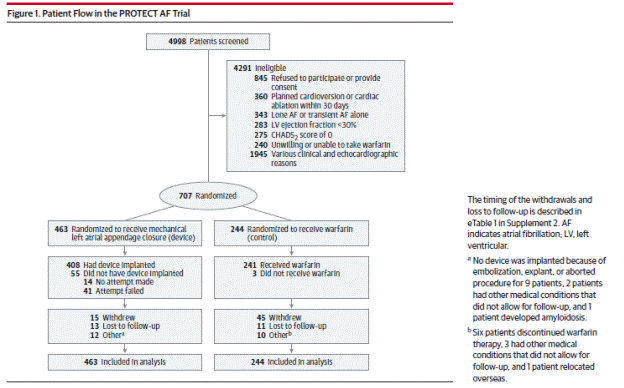
Page 1991. Figure 1. Patient flow in the PROTECT AF trial. Reddy VK et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation. A randomized clinical trial. JAMA 2014;312(19):1988-1998.
A higher proportion of patients in the warfarin control group, 22.5%, versus 6.3% in the device group, withdrew consent. The authors state that the principal reasons for withdrawal from the warfarin group were a desire for alternate treatment such as a NOAC and perceptions by patients in the warfarin group of little benefit from continued participation. The proportion of patients classified as lost to follow-up was similar in both groups (4.5% in the device group versus 2.8% in the warfarin group). Time in therapeutic window in the warfarin group was given as 70%.
In addition to an ITT analysis, the authors presented three additional patient population analyses. A post procedure analysis excluded patients in whom the device could not be successfully implanted and excluded events directly related to device implantation. A per -protocol analysis included device patients that had completed “the requisite 45-day period of anticoagulation after implantation.” A terminal therapy analysis assessed outcomes after device patients discontinued clopidogrel. As described previously, the primary analyses were based on ITT, censoring data from patients without events at the time of the last known status.
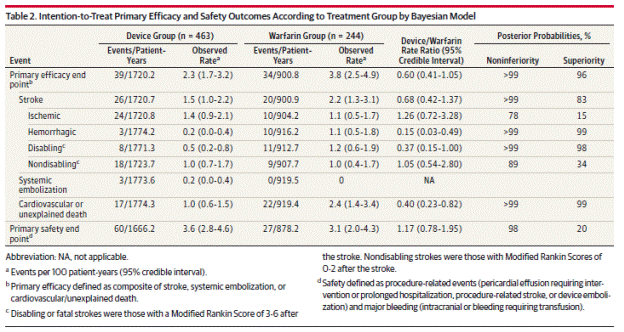
Page 1993. Table 2. Intention-to-treat primary efficacy and safety outcomes according to treatment group by Bayesian model. Reddy VK et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation. A randomized clinical trial. JAMA 2014;312(19):1988-1998.
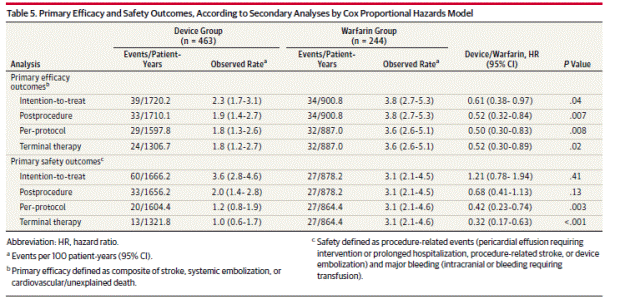
Page 1996. Table 5. Primary efficacy and safety outcomes, according to secondary analyses by Cox proportional hazards model. Reddy VK et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation. A randomized clinical trial. JAMA 2014;312(19):1988-1998.
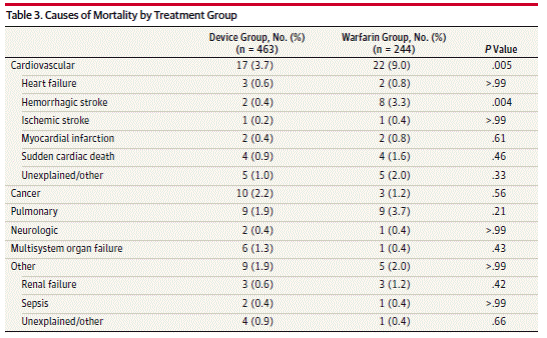
Page 1994. Table 3. Causes of mortality by treatment group. Reddy VK et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation. A randomized clinical trial. JAMA 2014;312(19):1988-1998. DOI:10.1001/jama.2014.15192
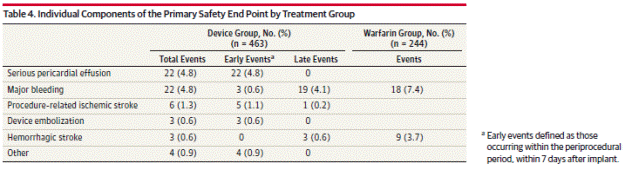
Page 1995. Table 4. Individual components of the primary safety end point by treatment group. Reddy VK et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation. A randomized clinical trial. JAMA 2014;312(19):1988-1998.
The authors note the rate of intracranial ICH in the warfarin groups (1.1% per year) is higher than in other trials, and that half the reported events occurred after falls.
The authors conclude, “After 3.8 years of follow-up in patients with Non-valvular AF at elevated risk for stroke, percutaneous LAA closure met criteria for both noninferiority and superiority, compared with warfarin therapy, for preventing the combined outcome of stroke, systemic embolism, and cardiovascular death, as well as superiority for cardiovascular mortality and all-cause mortality."
PREVAIL trial
Holmes DR, Kar D, Price MJ, Whisenant B, Sievert H, Doshi SK, Huber K, and Reddy VY. Prospective randomized evaluation of the WATCHMAN left atrial appendage closure device in patients with atrial fibrillation versus long-term warfarin therapy. The PREVAIL trial. J Am Coll Cardiol 2014;64:1-12.
This manufacturer-funded trial enrolled patients with NVAF and a CHADS2 of ≥2 or 1 and another risk factor (female age ≥75, baseline ejection fraction ≥ 30% but <35%, age 65 to 74 years and either diabetes or coronary disease, and age ≥65 years with congestive heart failure). This was done to include a higher risk group than patients in PROTECT AF. Similar to PROTECT AF, patients were randomized 2:1 stratified by center using a centralized computer system in block sizes of 6. This system was password protected and accessed by the principal investigator and study coordinator after the patient gave consent and met inclusion criteria. The study enrolled 475 patients with 407 through randomization (269 in the device arm and 138 in the control arm). New sites and new operators were included. The trial protocol was similar to PROTECT AF. Baseline demographic characteristics and risk factors for the randomized subjects were not statistically different between the device and control arm with one exception. The device arm had 238/269 (88.5%) of patients with a history of hypertension and the control arm had 134/138 (97.1%). The PREVAIL participants were higher risk on average than the PROTECT AF participants as demonstrated in the following table:
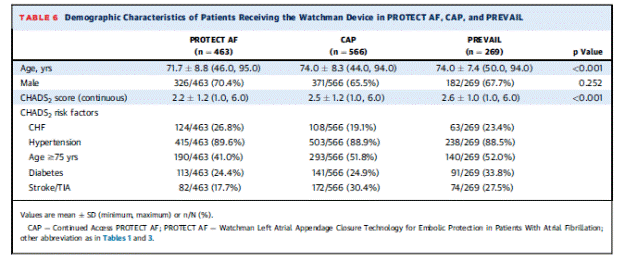
Page 4. Table 6. Demographic characteristics of patients receiving the WATCHMAN device in PROTECT AF, CAP, and PREVAIL. Holmes DR et al. Prospective randomized evaluation of the WATCHMAN left atrial appendage closure device in patients with atrial fibrillation versus long-term warfarin therapy. The PREVAIL trial. J Am Coll Cardiol 2014;64:1-12.
Three coprimary endpoints were defined. The first primary efficacy endpoint was the same as in PROTECT AF. The second coprimary efficacy endpoint was defined as ischemic stroke or SE > 7 days post-randomization. The third coprimary applicable only to the device arm was termed early safety, which included all-cause death, ischemic stroke, SE, or device-/procedure-related events requiring open cardiovascular surgery or major endovascular intervention between 7 days of the procedure or during the index hospitalization, with percutaneous catheter drainage of pericardial effusions, snaring of the embolized device, and nonsurgical treatments of access complications excluded. Statistical analysis is described in the article. The trial design was Bayesian adaptive. Data from PROTECT AF subjects that met the inclusion/exclusion criteria for PREVAIL used as an informative prior with a 50% discount. The rate ratio of 18-month event rates in the device and control groups were compared, with a risk ratio criterion of 1.75 to establish noninferiority for the first coprimary endpoint, as opposed to 2.0 in the PROTECT AF trial. The second coprimary was based on a null hypothesis that would be rejected if either the ratio or difference between rates in the groups satisfied the noninferiority criteria (<2.0 and <0.0275, respectively), without adjustment for multiple comparisons. The third coprimary had a performance goal for comparison.
Results for coprimary endpoints
For the randomized subjects, mean follow-up was 11.8 ±5.8 months, with a median of 12.0 months (range of 0.03 to 25.9 months).
- Endpoint of all stroke, systemic embolism, or cardiovascular/unexplained death
| Device 18-month rate |
Control 18-month rate |
18 month rate ratio (95%CrI) |
Rate ratio noninferiority criterion |
Noninferiority criterion definition met? |
| 0.064 |
0.063 |
1.07 (0.57,1.89) |
95% credible interval upper bound <1.75 |
No |
- Endpoint of stroke or SE>7 days after randomization for PREVAIL subjects only (ITT)
| Device 18-month rate |
Control 18-month rate |
18-month rate ratio (95% CrI) |
Rate ratio noninferiority criterion |
18-month rate difference (95% CrI) |
Rate difference noninferiority criterion |
Noninferiority criterion definition met? |
| 0.0253 |
0.0200 |
1.6 (0.5 to 4.2) |
95% CrI upper bound <2.0 |
00.53 (-0.0190 to 0.0273) |
95% CrI upper bound <0.0275 |
Yes (rate difference noninferiority) |
- Safety endpoint, device group only
- Safety primary endpoint, % (n/N): 2.2% (6/269) 95% CrI 2.652%
- Safety events by type:
- Device embolization 2
- Arteriovenous fistula 1
- Cardiac perforation 1
- Pericardial effusion with cardiac tamponade 1
- Major bleed requiring transfusion 1
The authors state that implantation success was achieved in 95.1% overall and in 96.3% with experienced operators versus 93.2% with new operators and no significant differences in complication rates.
The authors conclude, “This trial provides additional data that LAA occlusion is a reasonable alternative to warfarin therapy for stroke prevention inpatients with NVAF who do not have an absolute contraindication to short-term warfarin therapy.”
FDA Executive Summary Memorandum. Prepared for the December 11, 2013 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
PREVAIL
The PREVAIL trial was collaboratively designed by the manufacturer and the FDA to address major limitations from the PROTECT AF trial. This Bayesian adaptive study included a portion of the PROTECT AF data to be used as an informative prior. This panel meeting reviewed the results of PREVAIL and longer term follow-up data from PROTECT AF. PREVAIL changes in comparison to PROTECT AF:
Inclusion/exclusion criteria:
- Included subjects with CHADS2 score ≥ 2 or =1 if additional stroke risk factors were present and warfarin therapy was recommended according to 2006 AHA/ACC/ESC guidelines.
- Excluded subjects indicated for chronic clopidogrel therapy.
Analytic framework:
- For the first primary endpoint, noninferiority would be met if the upper bound of the equitailed 2-sided 95% CI for the 18 month rate ratio was less than 1.75.
- Addition of a second primary endpoint to compare the rate of ischemic stroke and SE beyond the first 7 days post-randomization to exclude peri-procedural events. Noninferiority would be met if the upper bound of the equitailed 2-sided 95% CI for the 18 month rate ratio was less than 2.0 or the upper bound of the equitailed 2-sided 95% CI for the 18 month rate difference was less than 0.0275.
- Inclusion of a third co-primary composite safety endpoint of occurrence of all-cause death, ischemic stroke, ER, or device or procedure-related events requiring open cardiac surgery or major endovascular intervention such as pseudoaneurysm repair, AV fistula repair, or other major endovascular repair, occurring between the time of randomization and within 7 days of the procedure or by hospital discharge, whichever is later.
Additionally, the manufacturer committed to improved enhanced monitoring of warfarin use and that 20% of subjects from new investigational sites and 25% of subjects implanted by new operators would be included.
Randomization, protocol description, and endpoint and statistical analysis are described in Holmes et al. 2014. Data collected in PROTECT AF and the CAP registry studies were utilized as prior information in both the design and analysis of the PREVAIL study. In the first and second primary endpoint analysis, only PROTECT AF subjects that would have met CHADS2 inclusion criteria were included, with a discounted weight of 50%. The third coprimary endpoint incorporated data from PROTECT AF and the CAP registry from patients with the same CHADS2 score inclusion as in PREVAIL. Subject accountability for PREVAIL is in Figure 2 of the FDA Summary. Subject demographics from Table 2 of the FDA Summary reveal a 94% Caucasian race/ethnicity. Baseline risk factors show a statistically significant difference in the percentage of subjects with a history of hypertension (88.5% in the device group vs. 97.1% in the control group, p = 0.003). The overall follow-up visit attendance was very high for both groups, between 96% and 100%. The duration of follow-up was 11.8 ± 5.8 months for the randomized subjects with only 28% of subjects having at least at 18 month follow-up visit.
The first primary endpoint based on the pre-specified hypothesis was not met. This 18-month rate is a model based rate of an event occurring within 18 months, as the rate is based on a piecewise exponential model and prior distributions constructed from the historical PROTECT AF trial data discounted 50%. The Kaplan-Meier estimate for the PREVAIL ITT population is Table 6 of the FDA Summary.
The second primary endpoint compared the rate ratio or the rate difference of the composite 18-month rate of stroke and SE excluding events occurring in the first 7 days. Similar to the first endpoint, this 18 month rate is a model based rate. The rate ratio noninferiority margin was not met. However, the noninferiority criteria was met based on the pre-specified hypothesis for the rate difference as endpoint noninferiority could be met based on either prespecified margin. The second primary endpoint events by type for the PREVAIL ITT population are in FDA Table 11 below:
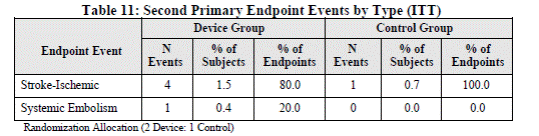
Page 27. Table 11: Second primary endpoint events by type (ITT). FDA Executive Summary Memorandum. Prepared for the December 11, 2013 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
The sponsor analyzed the first and second endpoints using only the PREVAIL data and the FDA analyzed the first and second endpoint using only PROTECT AF prior distribution data. The point estimates were different. The assumption of a constant hazard rate for each treatment group at particular follow-up intervals for the first and second primary endpoint analyses was evaluated based on the PREVAIL data and was found to be reasonable.
The third primary endpoint was a comparison to a performance goal: the composite rate of all-cause death, ischemic stroke, systemic embolism, or device- or procedure-related events requiring open cardiac surgery or major endovascular intervention between the time of randomization and within 7 days of the procedure or by hospital discharge, whichever was later. Events are listed in FDA Table 16. The event rate was 2.2%. The upper bound on the CI based on the Bayesian analysis was 2.652%, which met the performance goal of 2.67%. Two subjects who experienced the third primary endpoint event had additional adverse events:
- Patient had a cardiac arrest when the device became entangled in the mitral valve apparatus. The subject suffered anoxic encephalopathy and an ischemic stroke (captured in the first primary endpoint).
- Patient fell after implant and suffered a subdural hematoma that was adjudicated as a hemorrhagic stroke. The subject died 7 months later.
An analysis compared new operators (48%) to experienced operators (52%). Implantation by new operators was not associated with lower implant success or increased major adverse events.
Time in therapeutic range for the control group was 68%. Compliance with INR monitoring defined as an INR measurement taken at least every 28 days was 85%. Fifteen control subjects (10.9%) permanently discontinued warfarin. Of these 15, 7 switched to a NOAC, 3 had adverse events related to warfarin, 3 refused to continue warfarin therapy and 2 had LAA removal. Ninety-two percent of the device group discontinued warfarin 45 days post-procedure. Of the 19 that did not stop warfarin at 45 days, 5 continued warfarin due to jet size, and 3 had thrombus on the device found on TEE. The remainder continued on warfarin for various reasons as described in the FDA document. At 6 months post implant 98% of device subjects had discontinued warfarin.
Type and timing of major bleeding events is in Table 15 Appendix A. The FDA noted PREVAIL was not analyzed by bleeding scales typically used in other major clinical trials. There did not appear to be a signal of a reduction in total bleeding events or in events over time in the device group as compared to control.
There were 13 cases of device thrombus and none met the criteria for serious adverse events. Device recapture rate decreased significantly in PREVAIL compared to PROTECT AF.
On subgroup analysis, subjects with CHADS2 scores of 4-6 experienced a greater number of first and second primary endpoint events than subjects with CHADS2 scores of 1-3.
PROTECT AF
PROTECT AF data from the final pre-specified analysis at 1588 patient-years and a dataset at 2621 patient years, the most recent data, was reviewed. Subject withdrawal and lost to follow-up was not balanced between groups (FDA Table 37 below). A more detailed table for the 1588 patient years is FDA Summary Table 38.
Page 43. Table 37: PROTECT AF subject withdrawal and lost to follow-up. FDA Executive Summary Memorandum. Prepared for the December 11, 2013 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
The FDA comments that the rate of subject withdrawal, particularly the disparity in withdrawal rates between groups, could lead to bias against the control group and favor the device group for the long-term event rate comparisons, considering that the hazard rates decrease over time. The primary effectiveness endpoint was analyzed for both pt-yr datasets. However, the FDA commented that the model used to estimate the event rates was not accurate (assumption that the number of events followed a Poisson distribution with a constant hazard rate across the entire follow-up period within each treatment group, mentioned in the previous FDA summary) and reliance of the statistical results from the primary analysis was determined to be problematic. Events contributing to primary effectiveness endpoint for the ITT population at 2621 pt-yrs are in FDA Table 41. FDA Table 42 specifically examines ischemic stroke rates in the ITT populations and in a “post-procedure” population which included only device subjects with an implant attempt whereas the control group started from day of randomization. Based on these and similar analyses, the FDA concluded that after the procedural risk had been accounted for, the ischemic stroke rate in both treatment groups appeared to be similar. For the 2621 pt-yr analysis, the primary safety event rate was 3.6% for the device group and 3.1% for the control group. The number and rate of various types of safety events (provided by the sponsor) for both arms are FDA Table 47. As expected for an invasive procedure, device implantation had a higher rate of safety events. FDA Table 48 compares major bleeding results between arms. The FDA commented, “A postulated benefit of the WATCHMAN device compared with anticoagulation therapy is a reduced rate of bleeding events that would emerge in device subjects subsequent to the procedure and after warfarin was discontinued. Table 48 demonstrates a numerical trend consistent with this hypothesis. However, PROTECT AF was not designed or specifically powered to detect a difference in bleeding rates between WATCHMAN device and control subjects. The absence of a signal of reduced bleeding complications in PREVAIL should be considered in evaluation of [sic] the bleeding rates observed in PROTECT AF.”
Continued Access to PROTECT AF (CAP) Registry
The data included 566 subjects from 24 U.S. and 3 European sites. The inclusion and exclusion criteria and endpoints were the same as in the PROTECT AF trial. The follow-up schedule was similar to PROTECT AF except there was no requirement for TEE at 6 months if the LAA was determined to be closed at 45 days. Demographics were similar to PROTECT AF and are in FDA Table 50. Mean follow-up was 28.6 ± 10.6 months. About 20% of enrolled subjects had early exit from the study. The primary effectiveness event rate, in particular the ischemic stroke rate, was lower than the device group in PROTECT AF (FDA Table 53). No new safety issues were identified (FDA Table 56). The percentage of successfully implanted subjects who discontinued warfarin at 45 days post-procedure (95.8) was higher than that observed in PROTECT AF (86.8%) and PREVAIL (92%).
FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
The third FDA advisory panel meeting reviewed new data on device arm stroke rate in the PREVAIL trial. “Because the new ischemic strokes, all of which occurred in subjects who were more than one year post-WATCHMAN implant, raised concerns regarding the effectiveness of the device, FDA worked interactively with the sponsor to clarify the circumstances surrounding these new ischemic strokes. Responses to FDA’s questions were provided on May 2, 2014. In addition, enrollment in the CAP2 registry was suspended pending a complete analysis of the potential ischemic stroke signal in PREVAIL. The updated PREVAIL, CAP, and CAP2 data under review by the present Advisory Panel represents data locks as of June 28, 2014 (PREVAIL), June 25 2014 (CAP), and July 2, 2014 (CAP2).”
PREVAIL
FDA Table 1 provides a comparison of events for the previously reviewed dataset (FDA panel December 2013, data lock at January 2013) and the updated dataset (FDA panel October 2014, data lock at June 2014). These comments are noted:
- The ischemic stroke rate ratio of control vs. device is 0.15, p = 0.044 (FDA analysis)
- The ischemic stroke of SE rate ratio, control vs. device, is 0.14, p = 0.027 (FDA analysis)
- Hemorrhagic stokes were 2 events in each arm, rate ratio of control vs. device of 1.92, p = 0.61 (FDA analysis)
- “The rate of cardiovascular or unexplained death numerically favored the device group (rate ratio, control vs. device, is 1.45, p value = 0.575, FDA analysis), but none of the deaths was causally linked to the WATCHMAN device, implantation procedure, or anticoagulant therapy.”
Clinical narratives were reviewed to investigate the increasing number of events observed in the device group. Only one device event was related to the device implant procedure, where the device embolized and became entrapped in the mitral valve apparatus leading to open surgical removal and subsequent prolonged bypass time due to left ventricular dysfunction. Only one control subject experienced an ischemic stroke during this additional follow-up time, and it occurred when the INR was sub-therapeutic due to interruption for knee surgery. No mitigating circumstances were found to address concerns of device effectiveness.
The contribution of PREVAIL data to the primary and secondary endpoints is increased. The January 2013 analyses of PREVAIL data was heavily influenced by prior information from data borrowed from PROTECT AF with 50% discount (FDA Table 2). A comparison of the datasets for the first and second primary endpoint is below:
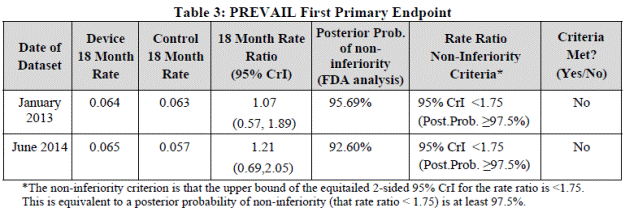
Page 15. Table 3: PREVAIL first primary endpoint. FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
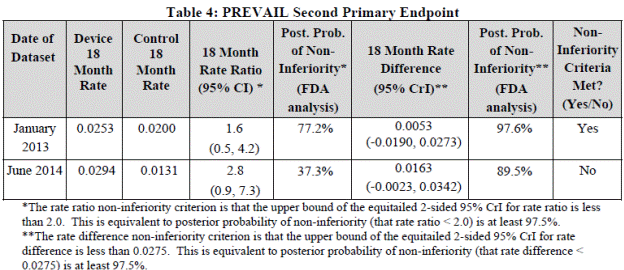
Page 16. Table 4: PREVAIL second primary endpoint. FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
The June 14 dataset for the first primary endpoint showed an increased difference in the 18 month rate ratio and the posterior probability of noninferiority was more distant from the success criterion. Similarly for the second primary endpoint, the risk ratio and risk difference for WATCHMAN vs. control was increased in favor of the control group, and the posterior probability of noninferiority was more distant from the success criterion. This change was driven by new ischemic strokes in the device group.
Further FDA analysis revealed that the PREVAIL and PROTECT AF distributions were divergent (Table 7, Table 8, Figure 2, Figure 3, Figure 4, and Figure 5 FDA Executive Summary). The FDA notes, “The reasons for the divergent clinical outcomes in PROTECT AF vs. PREVAIL are not clearly evident, but differences in trial conduct and execution may be contributory.”
The FDA analyzed the rate of ischemic stroke and SE by study group (Table 10, Table 11, Figure 6, Table 12, Table 13, Figure 7, Table 14, Table 15, and Table 16 FDA Executive Summary) and across all studies (Table 17 below).
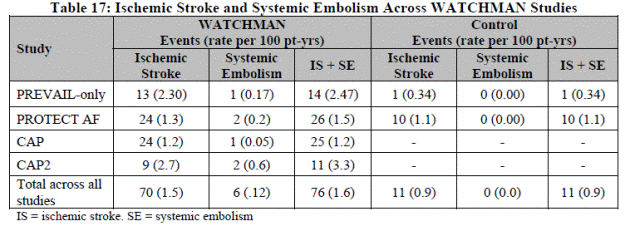
Page 29. Table 17: Ischemic stroke and systemic embolism across WATCHMAN studies. FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
In both PROTECT AF and PREVAIL the ischemic stroke plus SE rate favored the control group.
The hemorrhagic stroke rate across WATCHMAN studies is in Table 18 below. The following Table 20 compares hemorrhagic stroke rates in warfarin groups in contemporary anticoagulation trials.
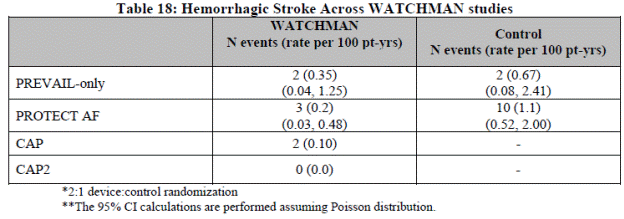
Page 30. Table 18: Hemorrhagic stroke across WATCHMAN studies. FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
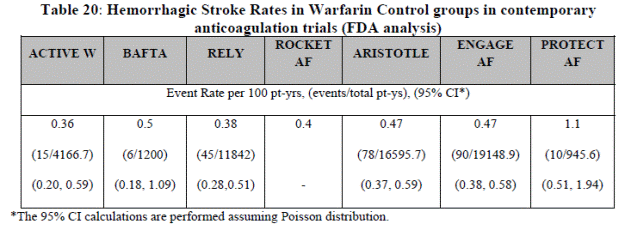
Page 31. Table 20: Hemorragic stroke rates in warfarin control groups in contemporary anticoagulation trials (FDA analysis). FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
The FDA reviewed the clinical narratives of the 10 hemorrhagic stroke cases in the PROTECT AF control group on warfarin therapy:
- One subject had been off warfarin for over 38 months and on aspirin alone.
- One subject did not have protocol required imaging confirmation. The clinical narrative included hypertension (210/100).
- Four subjects were on aspirin in addition to warfarin at the time of the stroke, and no antiplatelet use was available for another subject.
- Of the 10 events, 5 events occurred following falls. Four were associated with subdural hematomas and a subarachnoid hemorrhage was present in one subject. Three WATHCMAN subjects, two on aspirin alone and one on aspirin plus warfarin, fell and suffered subdural hematomas; these cases were not adjudicated as hemorrhagic strokes.
Major bleeding was defined as bleeding complications that were adjudicated as serious adverse events. Tables 21, 22, and 23 show the bleeding rates in PREVAIL, PROTECT AF, and CAP:
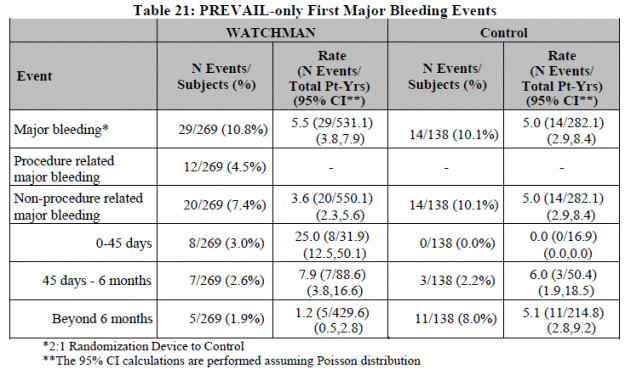
Page 34. Table 21: PREVAIL-only first major bleeding events. FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
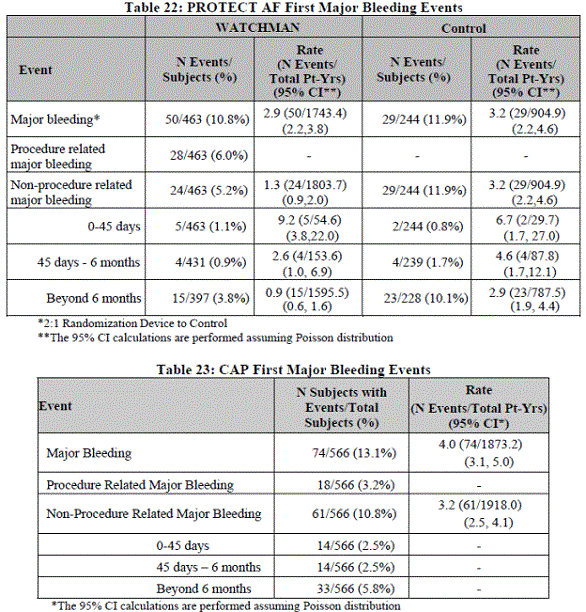
Page 35. Table 22: PROTECT AF first major bleeding events. Table 23: CAP first major bleeding events. FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
Cardiovascular and unexplained deaths were the third component of both the first primary endpoint in PREVAIL and the primary effectiveness endpoint in PROTECT AF. Comparison across studies including the registries are below:
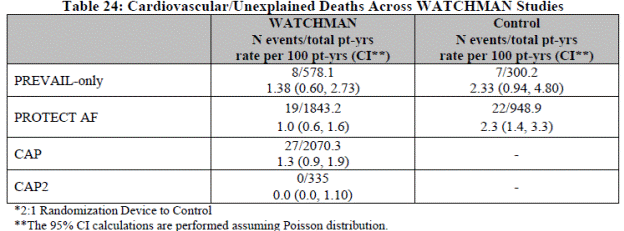
Page 36. Table 24: Cardiovascular/unexplained deaths across WATCHMAN studies. FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
In PROTECT AF, of the 22 control group cardiovascular or unexplained deaths, 8 occurred in subjects adjudicated as having hemorrhagic stroke. The FDA notes that in PREVAIL, the rate favors the device group, but none of the deaths were causally lined to the WATCHMAN device, implantation procedure, or anticoagulant therapy.
All-cause mortality across WATCHMAN studies is presented in Table 25 in the FDA Summary. The FDA comments that mortality rate inferences are limited by underlying subject co-morbidities where the modes of death in most subjects had little relationship to the device or anticoagulation.
The FDA evaluated the performance of the warfarin control groups compared to warfarin treatment in anticoagulation drug trials (Table 26 and 27 FDA Executive Summary). Time in therapeutic range (TTR) is the standard for reporting quality of INR control and warfarin dosing, i.e., the more the time in TTR the better the subject outcome. Figure 8 below provides a trend line:
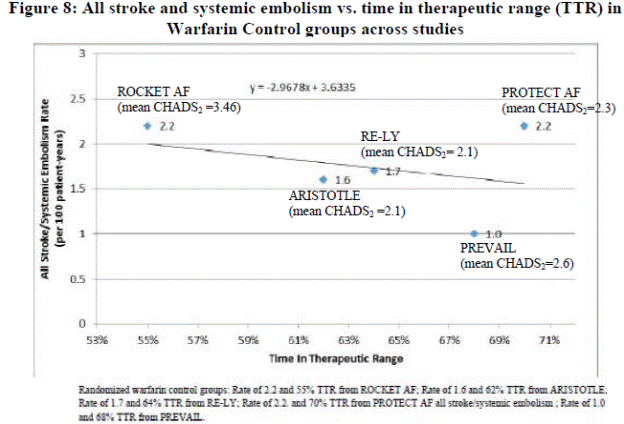
Page 39. Figure 8. All stroke and systemic embolism vs. time in therapeutic range (TTR) in warfarin control groups across studies. FDA Executive Summary Memorandum. Prepared for the October 8, 2014 meeting of the Circulatory System Devices Advisory Panel. P130013. Boston Scientific WATCHMAN® Left Atrial Appendage Closure Therapy.
Holmes DR, Doshi SK, Kar S, Price MJ, Sanchez JM., Sievert H, Valderrabano M, Reddy VY. Left atrial appendage closure as an alternative to warfarin for stroke prevention in atrial fibrillation. A patient-level meta-analysis. J Am Coll Cardiol 2015;65:2614-23.
This study combines data from PROTECT AF, CAP registry, PREVAIL, and the CAP2 registry. The authors report the mean follow-up for the entire study population was 2.69 years to report on 2,406 patients for a total of 5,931 pt-yrs.
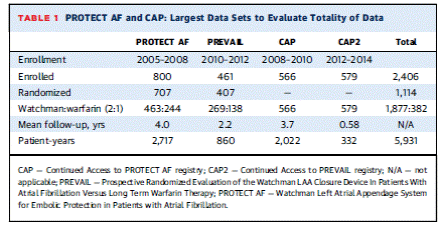
Page 2618. Table 1. PROTECT AF and CAP: largest data sets to evaluate totality of data. Holmes DR et al. Left atrial appendage closure as an alternative to warfarin for stroke prevention in atrial fibrillation. A patient-level meta-analysis. J Am Coll Cardiol 2015;65:2614-23.
The figure below illustrates the results only from the combined two RCTs:
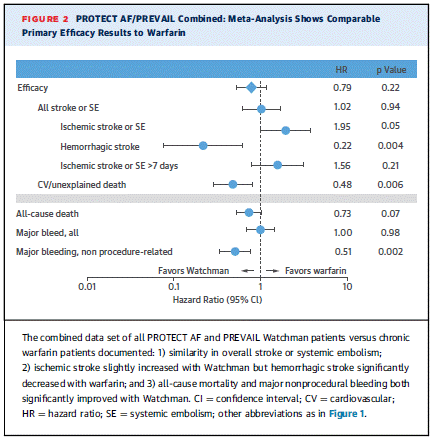
Page 2620. Figure 2. PROTECT AF/PREVAIL combined meta-analysis shows comparable primary efficacy results to warfarin. Holmes DR et al. Left atrial appendage closure as an alternative to warfarin for stroke prevention in atrial fibrillation. A patient-level meta-analysis. J Am Coll Cardiol 2015;65:2614-23. Doi: 10/1016/j.jacc.2015.04.025
The authors conclude, "In patients with NVAF at increased risk for stroke or bleeding who are candidates for chronic anticoagulation, LAAC results in improved rates of hemorrhagic stroke, cardiovascular/unexplained death, and nonprocedural bleeding compared to warfarin."
Case Series in Patients with a Contraindication to Oral Anticoagulants
Reddy VY, Mobius-Winkler S, Miller M, Neuzil P, Schuler G, Wiebe J, Sick P, Sievert H. Left atrial appendage closure with the WATCHMAN device in patients with a contraindication for oral anticoagulation: ASA Plavix feasibility study with WATCHMAN left atrial appendage closure technology (ASAP Study). J Am Coll Cardiol 2013. Jun 25;61(25):2551-6.
The purpose of this study was to assess safety and efficacy of the WATCHMAN device in patients determined to be ineligible for warfarin therapy. One hundred fifty patients with NVAF were enrolled in 4 centers (3 OUS) between January 2009 and November 2011. Included criteria were CHADS2 ≥ 1 and a contraindication for even short term OAC therapy (hemorrhagic/bleeding tendency – defined as active peptic ulcer disease or history of overt bleeding of the gastrointestinal, genitourinary, or respiratory tract; central nervous system hemorrhage, cerebral aneurysm, dissection of the aorta, pericarditis/pericardial effusions or bacterial endocarditis; blood dyscrasias; unsupervised patients with senility and/or high fall risk; and other documented reasons) but eligibility for antiplatelet agents. Patients only received antiplatelet agents after WATCHMAN implantation. The primary efficacy endpoint was similar to PROTECT AF. A serious adverse event was defined as a medical condition that results in death, was life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, or resulted in persistent or significant disability/incapacity. Clinical characteristics of the study group included a mean age of 72.5 ± 7.4, 94.7% with hypertension, mean CHADS2 score of 2.8 ± 1.2, and 93% with a history of hemorrhagic/bleeding tendencies. Mean duration of follow-up was 14.4 ± 8.6 months. Thirteen subjects (8.7%) had procedure or device related serious adverse events. There were 6 cases on device-related thrombus with only one adjudicated (341 days post implant) as a serious adverse event. There were 176.9 pt-yrs of follow-up. Clinical outcomes included 3 ischemic strokes, 1 hemorrhagic stroke, and 9 deaths. Primary efficacy events were 8 per 175.0 pt-yrs, or 4.6%. The authors conclude, “LAA closure with the WATCHMAN device can be safely performed without a warfarin transition, and is a reasonable alternative to consider for patients at high risk for stroke but with contraindications to systemic oral anticoagulation.”
Matsuo Y, Sandri M, Mangner N, Majunke N, Dahnert I, Schuler G, Kurabayashi M, Mobius-Winkler S. Interventional closure of the left atrial appendage for stroke prevention. Experience of a high-volume center. Circ J 2014; 78:619-624.
The authors report on a case series of 179 patients who underwent WATCHMAN implantation and 11 patients who underwent Amplatzer implantation by two experienced investigators at a single center in Germany. This study was a safety and feasibility study of patients over 18 years with documented chronic or paroxysmal NVAF and an estimated life expectancy of at least 2 years. Included patients had a relative contraindication to OACs such as bleeding history, renal dysfunction, liver dysfunction, high risk of falling or drug non-compliance. Exclusion criteria were symptomatic valvular disease , symptomatic carotid disease and pregnancy as well as an indication other than AF for OAC therapy. Patients either remained on warfarin for a minimum of 45 days or had therapy with dabigatran, dual antiplatelet, or enoxaparin. If criteria for LAA sealing were met then warfarin/dabigatran or enoxaparin were discontinued while aspirin plus clopidogrel were continued for 4 to 5 months followed by aspirin indefinitely. Follow-up was reported up to 6 months. Baseline patient characteristics, include a HAS-BLED score of 3.9 ±1.1 and 39.8% with CHADS2 ≤ 2. The overall procedural complication rate was 11.2%. A TIA occurred in 2 patients during 6 month follow-up. No cases of stroke were reported. One patient had a traumatic subdural hematoma. Nine patients were found to have thrombus on the device without embolization. The authors conclude, “Transcatheter LAA closure in a high-volume center is safe and feasible. Life-threatening complications are rare. Discontinuation of OAC 45 days after implantation was also safe.”
Nietlispach F, Gloekler S, Krause R, Shakir S, Schmid M, Khattab AA, Wenaweser P, Windecker S, and Meier B. Amplatzer left atrial appendage occlusion: single center 10-year experience. Catheter Cardiovasc Interv. 2013 Aug 1;82(2):283-9.
The authors report outcomes for 152 patients from a single center in Switzerland with various devices including the Amplatzer Cardiac Plug (ACP), a device that is FDA approved for atrial septal defect closure but not for LAA occlusion. Inclusion criteria included CHA2DS2-VASc score of ≥ 1, and a history of previous bleeding, patients at high-risk of bleeding, or patient preference. The safety endpoint was defined as the composite of procedure-related complications (pericardial effusion, device embolization, and procedure-related stroke) and major bleeds (according to TIMI Bleeding Criteria for classification of Non-CABG related bleeding). The efficacy endpoint was defined as the composite of cardiovascular and unexplained deaths, neurologic events (ischemic and hemorrhagic stroke, TIAs), and SE. Patients were discharged on dual antiplatelets for 1-6 months unless they had concomitant left atrial ablation for which they remained on OAC for 3 months. Clinical characteristics of the overall population included mean CHA2DS2-VASc score of 3.4 ± 1.7, and a HAS-BLED score of 2.4 ± 1.2. Mean follow-up was 32 months (range 1 – 120). The authors report that the composite efficacy endpoint occurred in 11 patients (7.2%) and the safety endpoint occurred in 19 patients (12.5%):
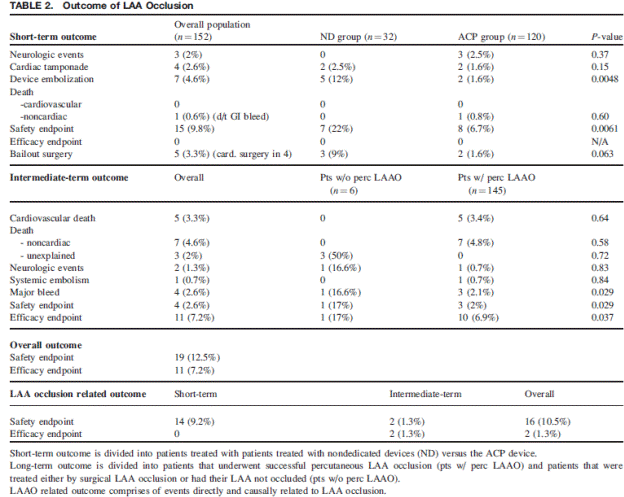
Table 2. Outcome of LAA occlusion. Page 287. Nietlispach F et al. Amplatzer left atrial appendage occlusion: single center 10-year experience. Catheter Cardiovasc Interv. 2013 Aug 1;82(2):283-9
The authors conclude, “LAA closure may be a good alternative to oral anticoagulation. This hypothesis needs to be tested in a randomized clinical trial to ensure that all potential biases of the observational study are accounted for.”
Wiebe J, Bertog S, Franke J, Wettstein O, Lehn K, Hofmann I, Vaskelyte L, and Sievert H. Safety of percutaneous left atrial appendage closure with the Amplatzer Cardiac Plug in patients with atrial fibrillation and contraindications to anticoagulation. Catheter Cardiovasc Interv. 2014 Apr 1;83(5):796-802.
The stated purpose of this study was to evaluate the safety of left atrial appendage closure with the ACP in patients with NVAF who are not eligible for warfarin. Between January 2009 and December 2012 sixty consecutive patients with a CHADS2 score of ≥1 and who were not eligible for warfarin were analyzed retrospectively. The authors state, “This includes patients with contraindications as defined in the warfarin product label, a history of severe bleeding while receiving anticoagulant therapy as well as a history of bleeding tendencies in the absence of anticoagulation of blood dyscrasia. Patients who were unable to maintain a stable INR and those with a known hypersensitivity to warfarin or a high-risk of falling were also included. Patients who requested elective LAA closure despite an absence of contraindications to anticoagulation were not enrolled in this study.” Study population characteristics included a mean CHADS2 score 2.6 ± 1.4, and a HAS-BLED score of 3.3 ± 1.0. Post-procedure medical therapy included life-long aspirin and 3 months of clopidogrel. Mean follow-up time was 1.8 (1.0-2.8) years for a total of 103.2 pt-yrs. Implantation success was 95.0%. There were no reported ischemic strokes or TIAS, or any other thromboembolic events during the follow-up time. Major adverse events were defined as events that were fatal, life-threatening, needed hospitalization, or prolongation of current hospitalization or led to considerable disability. Seven patients had a procedure related AE (2 device embolizations, 2 transient ST elevations, one pericardial effusion, one hematoma, one allergic reaction). There were 5 deaths and 9 bleeding complications, of which 2 were major. One was a traumatic intracranial hemorrhage. The authors conclude, “Percutaneous LAA closure with the ACP in patients with contraindications to oral anticoagulation is safe. The stroke and bleeding risk after percutaneous LAA closure is lower than predicted by conventional risk scores.”
4. Medicare Evidence Development and Coverage Advisory Committee (MEDCAC)
A MEDCAC meeting was not convened on this issue.
5. Evidence-based guidelines
You JJ, Singer DE, Howard PA, Lane DA, Eckman MH, Fang MC, Hylek EM, Schulman S, Go AS, Hughes M, Spencer FA, Manning WJ, Halperin JL, Lip GYH. Antithrombotic Therapy for Atrial Fibrillation. Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-based clinical practice guidelines. Chest 2012; 141(2)(Suppl):e531S-e575S.
“For patients with nonrheumatic AF, including those with paroxysmal AF, who are (1) at low risk of stroke (eg, CHADS2 [congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke or transient ischemic attack] score of 0), we suggest no therapy rather than antithrombotic therapy, and for patients choosing antithrombotic therapy, we suggest aspirin rather than oral anticoagulation or combination therapy with aspirin and clopidogrel; (2) at intermediate risk of stroke (eg, CHADS2 score of 1), we recommend oral anticoagulation rather than no therapy, and we suggest oral anticoagulation rather than aspirin or combination therapy with aspirin and clopidogrel; and (3) at high risk of stroke (eg, CHADS2 score of ≥ 2), we recommend oral anticoagulation rather than no therapy, aspirin, or combination therapy with aspirin and clopidogrel. Where we recommend or suggest in favor of oral anticoagulation, we suggest dabigatran 150 mg bid rather than adjusted-dose vitamin K antagonist therapy.”
“At this time, we make no formal recommendations regarding LAA closure devices, pending more definitive research in the field.”
Meschia JF, Bushnell C, Boden-Albala B, Braun LT, Bravata DM, Chaturvedi S, Creager MA, Eckel RH, Elkind MSV, Fornage M, Goldstein LB, Greenberg SM, Horvath SE, Iadecola C, Jauch EC, Moore WS, Wilson JA; on behalf of the American Heart Association Stroke Council, Council on Cardiovascular and Stroke Nursing, Council on Clinical Cardiology, Council on Functional Genomics and Translational Biology, and Council on Hypertension. Guidelines for the primary prevention of stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:3754–3832.
“Closure of the LAA has been evaluated as an alternative approach to stroke prevention in Non-valvular AF. In a trial of 707 subjects randomized 2:1 to percutaneous LAA closure with the WATCHMAN device (in which patients were treated with warfarin for at least 45 days after device placement, then aspirin plus clopidogrel from echocardiographically demonstrated closure of the LAA until 6 months after placement, then aspirin alone) versus adjusted-dose warfarin (target INR, 2 to 3), LAA closure was noninferior to warfarin for preventing the primary outcome of ischemic or hemorrhagic stroke, cardiac or unexplained death, or systemic embolism during the mean 18-month follow-up (RR, 0.62; 95% CI, 0.35–1.25; P<0.001 for noninferiority). Hemorrhagic stroke was less frequent in the LAA closure group (RR, 0.09; 95% CI, 0–0.45), but ischemic stroke was insignificantly more frequent (RR, 1.34; 95% CI, 0.60–4.29), in part because of procedure-related strokes (occurring in 5 of the 449 patients in whom LAA closure was attempted, including 2 with longterm residual deficits). At 1588 patient-years of follow-up, the rate of the primary efficacy end point of stroke, systemic embolism, and cardiovascular death was not inferior for the WATCHMAN device compared with warfarin. Although this approach appears promising, there are substantial reasons for proceeding cautiously with this treatment, including the relatively modest power of the trial, the exclusion of subjects with firm contraindications to anticoagulation (who would otherwise appear to be ideal candidates for LAA closure), and the lack of comparison to the newer, potentially more effective oral anticoagulants.”
Recommendations
Closure of the LAA may be considered for high-risk patients with AF who are deemed unsuitable for anticoagulation if performed at a center with low rates of periprocedural complications and the patient can tolerate the risk of at least 45 days of postprocedural anticoagulation (Class IIb; Level of Evidence B).[1]
Culebras A, Messe SR, Chaturvedi S, Kase C, and Gronseth G. Summary of evidence-based guideline update: prevention of stroke in Non-valvular atrial fibrillation. Report of the guideline development subcommittee of the American Academy of neurology. Neurology 2104;82:716-724.
“Selection of patients for antithrombotic therapy. Clinical context. Within the NVAF of other stroke risk factors. The absolute stroke risk is highest among patients with NVAF and a history of stroke and TIA (aggregated absolute risk about 10%/y). Although multiple risk stratification tools are available for estimating the absolute stroke risk of patients with NVAF, the absolute stroke risks estimated by these tools vary widely.
Because it is difficult to determine with precision the absolute stroke risk in patients with NVAF, determining when the benefit from reduced stroke risk outweighs the harm of increased bleeding is likewise difficult. In these circumstances, patient preferences and physician judgment become especially important.”
Practice recommendations
B1. Clinicians should inform patients with NVAF that these patients have an increased stroke risk and that this risk can potentially be reduced by antithrombotic use. Patients should also be informed that antithrombotic use increases their risk of major bleeding (Level B).
B2. Clinicians should counsel all patients with NVAF that the decision to use antithrombotics must be made only after the potential benefit from the stroke risk reduction has been weighed against the potential harm from the increased risk of major bleeding. Clinicians should also emphasize the important role of judgment and preferences in this decision (Level B).
B3. Clinicians should routinely offer anticoagulation to patients with NVAF and a history of TIA or stroke, to reduce these patients’ subsequent risk of ischemic stroke (Level B).
B4. Clinicians might not offer anticoagulation to patients with NVAF who lack additional risk factors (“lone” NVAF patients). Clinicians might reasonably offer antithrombotic therapy with aspirin to such patients or might not offer antithrombotic therapy at all (Level C).
B5. To inform their judgments as to which patients with NVAF might benefit more from anticoagulation, clinicians should use a risk stratification scheme to help identify patients with NVAF who are at higher risk for stroke or at no clinically significant risk. However, clinicians should not rigidly interpret anticoagulation thresholds suggested by these tools as being definitive indicators of which patients require anticoagulation (Level B).”
National Clinical Guideline Centre (UK)
Atrial Fibrillation: The Management of Atrial Fibrillation.
London: National Institute for Health and Care Excellence (UK); 2014 Jun.
“Consider left atrial appendage occlusion (LAAO) if anticoagulation is contraindicated or not tolerated and discuss the benefits and risks of LAAS with the person.” “Do not offer LAAO as an alternative to anticoagulation unless anticoagulation is contraindicated or not tolerated.”
The suggested algorithm for stroke prevention in people with NVAF:
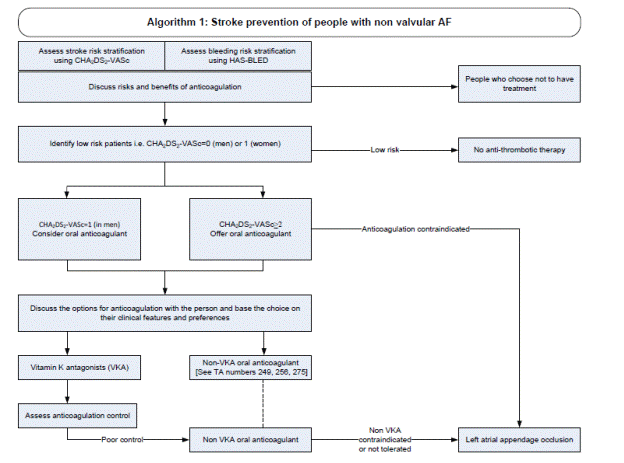
Page 42. Algorithm 1: Stroke prevention of people with non valvular AF. National Clinical Guideline Centre. Atrial Fibrillation: the management of atrial fibrillation. Clinical Guideline. Methods, evidence, and recommendations. June 2014.
Verma A, Cairns JA, Mitchell LB, Macle L, Stiell IG, Gladstone D, McMurtry MS, Connolly S, Cox JL, Dorian P, Ivers N, Leblanc K, Nattel S, Healey JS; for the CCS atrial fibrillation guidelines committee. 2014 focused update of the Canadian Cardiovasuclar Society guidelines for the management of atrial fibrillation. Can J Cardio. 2014;30(10):1114-1130.
Left atrial appendage closure in stroke prevention
“We suggest these nonapproved LAA closure devices not be used, except in research protocols or in systematically documented use protocols in patients at high risk of stroke (CHADS2 score ≥ 2) for whom antithrombotic therapy is precluded (Conditional Recommendation, Low-Quality Evidence).”
Camm AJ, Lip GYH, De Caterina R, Savelieva I, Atar D, Hohnloser SH, Hindricks G, Kirchhof P. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation. An update of the 2010 ESC Guidelines for the management of atrial fibrillation. Europace. 2012;14(10):1382-1413.
“Interventional, percutaneous LAA closure may be considered in patients with a high stroke risk and contraindications for long-term oral anticoagulation.” Class of recommendation IIb; Level of evidence B.
6. Consensus Statements
Masoudi FA, Calkins H, Kavinsky CJ, Drozda JP Jr, Gainsley P, Slotwiner DJ, Turi ZG. 2015 ACC/HRS/SCAI left atrial appendage occlusion device societal overview: a professional society overview from the American college of Cardiology, Heart Rhythm Society, and Society for Cardiovascular angiography and Interventions. J Am Coll Cardiol. 2015.
The purpose of this document is “to highlight the critical issues surrounding LAA occlusion therapies and to facilitate the alignment of multiple interests...” Recommendations:
Care team and facilities
Multidisciplinary Heart Team
- “The initial evaluation should be performed by both an individual with the expertise to characterize the specific risks and benefits of medical therapy and a procedural specialist, who can estimate the risks and benefits of a proposed procedure.”
- “The procedural specialist should also have expertise related to medical therapy for stroke prevention in AF.”
- “Beyond the initial evaluation, any input and/or participation in evaluating and managing this procedure should include expertise in echocardiography, x-ray imaging modalities (primarily computed tomography [CT]), and in anesthesiology when general anesthesia is planned.”
- “A cardiac surgeon should be available for surgical backup in case of emergency.”
General requirements
- “One of the cornerstones of a structural heart disease and/or electrophysiology program is a well-formulated, collaborative effort among all members of the care team. Depending on the type of procedure and device use, close collaboration may be required between procedural specials, physician echocardiographers, sonographers, radiologists, hematologists, neurologists, and cardiac surgeons to ensure proper patient selection, evaluation, and execution of LAA occlusion. In some cases, expertise in other areas may be required to inform decision making (e.g., geriatric medicine and/or gastroenterology or urology for patients with a history of significant gastrointestinal or genitourinary bleeding, respectively).”
- “Irrespective of specialty, physicians performing these procedures should possess the appropriate cognitive and technical skillsets. They should have an understanding of stroke and stroke syndromes, AF, the pharmacology of anticoagulants, and the regional anatomy of the left atrium and LAA.”
- “LAA occlusion procedures are complex and should be performed in institutions with experience in advanced structural heart disease procedures and/or electrophysiology procedures that require access to the left atrium.”
- “Collectively, members of the multidisciplinary heart team must be skilled in imaging of the LAA, trans-septal techniques, percutaneous pericardial puncture, advanced retrieval techniques, and large vessel access.”
- “Although the minimum training for these procedures may initially be prescribed by FDA approval requirements, competence in atrial septal puncture and proper handling of device inside of the left atrium to prevent air embolization and clot formation are prerequisites.”
- “The team should be structured to permit the consideration of all of the available therapeutic options to the patient individualized to the risks and benefits of these approaches on the basis of available data.”
Facilities
“The institution should have an established structural heart disease and/or electrophysiology program with an individual capable of performing the procedure as well as cardiac surgical backup. The full range of facilities for diagnostic imaging as well as electrophysiology, interventional, or cardiac surgical suites should be available on site and should include the following personnel and equipment:
- A cardiac procedure laboratory (electrophysiology or cardiac catheterization) or hybrid operating room equipped with a radiographic imaging system with fluoroscopy offering catheterization-quality imaging. A biplane unit may be useful in LAA occlusion procedures but is not required. Continuous hemodynamic monitoring is required during the procedure.
- An echocardiographic laboratory with the full array of transthoracic and transesophageal capabilities. Three-dimensional and intracardiac echocardiography or intracardiac echocardiography may be useful but are not required. A transesophageal echocardiogram-capable machine should be utilized during the case. Appropriate staff should be present, including a physician echocardiographer skilled in the subtleties of the procedure and available throughout the case.
- A CT laboratory with CT technologists and specialists skilled in obtaining high-quality cardiac studies of the heart for procedures where CT imaging is necessary as part of the evaluation (e.g., LARIAT). Preferable CT studies would be gated to optimize image resolution.
- A cardiac surgeon and anesthesiologist on site available for surgical backup.
- Cardiac surgery operating rooms in reasonable proximity to the room in which the procedure is being performed and readily accessible.
- A room of sufficient size to accommodate all of the necessary equipment and personnel.
- The full array of equipment necessary to conduct structural heart disease interventions and device retrieval within the procedural suite.
- An intensive care facility with staff trained to provide postprocedural observation and management.”
Operator Training
- “The ACC, HRS, and Society for Cardiovascular Angiography and Interventions have published recommendations for training in electrophysiology or interventional techniques, but these recommendations do not provide specific guidance for LAA occlusion devices. Thus, specific recommendations for training in LAA occlusion need to be developed.”
Protocols for Care
- “Specific protocols for preprocedural, intraprocedural, and postprocedural patient assessment and care should be in place, with clear delineation of the
roles of heart team members and the specific collaborative process for shared decision making with the patient.”
- “Protocols should involve assessment of the following: the patient’s’ stroke risk (preferably using the CHA2DS2 -VASc score), bleeding risk (using a bleeding risk score), any contraindications to anticoagulation, patient adherence to and history of adequacy of anticoagulation, cardiac structural factors (left ventricular ejection fraction and the presence of structural abnormalities such as patent foramen ovale, interatrial septal aneurysm, and LAA thrombus), and patient preferences.”
- “Documentation should include the decision making involved, including the consideration of pharmacotherapy as an alternative.”
- “Protocols are also needed to standardize preprocedural evaluation, including a complete assessment of medical comorbidities, preprocedural and inraprocedural imaging, and surgical backup.”
- “Finally, protocols are also needed to standardize postprocedure evaluation and follow-up.”
Assessment of patient selection and outcomes
- “The addition of LAA occlusion to an existing registry or creation of a new registry similar to the TVT registry would greatly benefit the still-nascent field of LAA occlusion technologies, in which the use of only 1 of multiple technologies is supported by randomized control trial data and significant learning curve effects exist.”
Meier B, Blaauw Y, Khattab AA, Lewalter T, Sievert H, Tondo C, Glikson M; Document reviewers: Lip GYH, Lopez-Minguez J, Roffi M, Israel C, Dudek D, Savelieva I. EHRA/EAPCI expert consensus statement on catheter-based left atrial appendage occlusion. Europace 2014;16(10)1397-1416.
The recommended indications for LAA occluders are summarized in this algorithm:
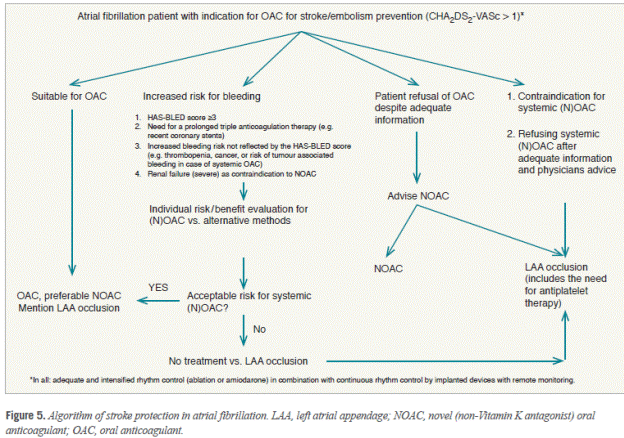
Page 1409. Atrial fibrillation patient with indication for OAC for stroke/embolism presentation (CHA2DS2-VASc > 1). Meier B et al. EHRA/EAPCI expert consensus statement on catheter-based left atrial appendage occlusion. Europace 2014;16(10)1397-1416.
7. Public Comments
Initial Comment Period: 5/20/2015 – 6/21/2015
During the initial 30-day comment period, CMS received 89 comments (90 comments were actually received but one comment was submitted in duplicate). CMS received the majority of comments from cardiac electrophysiologists (29/89), interventional cardiologists (13/89), and general cardiologists (10/89). Two comments were submitted by joint professional societies, one from the AHA/ASA and the other by ACC/HRS/SCAI. These professional societies expressed the need for further studies and limited coverage through CED. They included suggested CED criteria to be considered. Seven individuals did not identify an associated organization or profession; one comment was submitted by a device manufacture and the remaining 27 comments were submitted by physicians; research coordinators; directors of cardiac catherization labs; professors of medicine; consultants and directors of cardiovascular medical centers. The comments were generally supportive of coverage with several commenters advocating for coverage for percutaneous LAAC therapy with restrictions.
The comments can be viewed in their entirety on our website at https://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=281.
VIII. CMS Analysis
National coverage determinations (NCDs) are determinations by the Secretary with respect to whether or not a particular item or service is covered nationally by Medicare (§1869(f)(1)(B) of the Act). In order to be covered by Medicare, an item or service must fall within one or more benefit categories contained within Part A or Part B, and must not be otherwise excluded from coverage. Moreover, with limited exceptions, the expenses incurred for items or services must be "reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member." See §1862(a)(1)(A)of the Social Security Act.
In addition to § 1862(a)(1)(A) of the Act, a second statutory provision may permit Medicare payment for items and services in some circumstances. That statute, section 1862(a)(1)(E) of the Act, provides, in pertinent part, that:
(a) Notwithstanding any other provision of this title, no payment may be made under part A or part B for any expenses incurred for items or services—
. . .
(1)(E) in the case of research conducted pursuant to section 1142, which is not reasonable and necessary to carry out the purposes of that section[.]
Section 1142 of the Act describes the authority of the Agency for Healthcare Research and Quality (AHRQ) to conduct and support research on outcomes, effectiveness, and appropriateness of services and procedures to identify the most effective and appropriate means to prevent, diagnose, treat, and manage diseases, disorders, and other health conditions. That section includes a requirement that the Secretary assure that AHRQ research priorities under Section 1142 appropriately reflect the needs and priorities of the Medicare program.
CED is a paradigm whereby Medicare covers items and services on the condition that they are furnished in the context of approved clinical studies or with the collection of additional clinical data. In making coverage decisions involving CED, CMS decides after a formal review of the medical literature to cover an item or service only in the context of an approved clinical study or when additional clinical data are collected to assess the appropriateness of an item or service for use with a particular beneficiary.
As noted earlier, our review sought answers to the questions below. We have repeated them here for the convenience of the reader.
Question(s)
Is the evidence sufficient to conclude that percutaneous left atrial appendage closure (LAAC) improves health outcomes for Medicare beneficiaries with non-valvular atrial fibrillation?
If the answer to the questions above is positive, is the available evidence sufficient to identify the characteristics of the patient, practitioner or facility that predict which beneficiaries are more likely to experience overall benefit or harm from LAAC?
Analysis
Stroke is a leading cause of death and functional impairment. There is a positive trend as mortality rates and incidence have been declining for decades. This downward trend has occurred in tandem with cardiovascular risk factor control interventions such as hypertension control and smoking cessation programs. A large international case-control study of ischemic and hemorrhagic stroke concluded, “Targeted interventions that reduce blood pressure and smoking, and promote physical activity and a healthy diet, could substantially reduce the global burden of stroke” (O’Donnell et al. 2010). Effective prevention is the best approach for reducing the burden of stroke. Numerous factors, both modifiable such as hypertension and non-modifiable such as age, contribute to risk. Stroke risk is complex and not completely understood, however it is understood that effective measures such as blood pressure reduction and smoking cessation reduce the incidence of stroke.
AF is the most common sustained heart rhythm disturbance and the incidence increases with age. AF appears to have various etiologies, including many conditions other than traditional heart disease, though understanding of mechanisms and etiology remain incomplete (Wyse et al. 2014). The annual incidence of AF per 1000 person-years is 1.9 in women and 3.1 in men younger than 65 years, then exceeds 32 per 1000 person-years in patients 80 years and older (Piccini et al. 2012). AF is a major risk factor for cardioembolic stroke, but not all strokes that occur in elderly, frequently hypertensive AF patients are caused by cardioembolism from the LA/LAA. Strokes in these patients may be caused by cerebrovascular disease, complex atheromatous plaques involving the aorta and carotid arteries, and other cardiac sources (Hart et al. 2000; Arboix and Alió 2010; Wrigley and Lip 2009). An estimated 15% of all strokes are caused by AF, with a significant increase in percentage with age (Lip and Lim 2007). In the Framingham cohort, 23.5% of strokes were felt to occur as a direct result of AF in 80-89 year olds (Wolf et al. 1991). In a large community based study of patients with stroke with case ascertainment by CT or necropsy to determine the pathological type, the prevalence of AF was 17% for all stroke types, with the highest prevalence in large infarcts presumed to be due to occlusion of the main stem of the middle cerebral artery and lowest in patients with lacunar infarction (Sandercock et al. 1992). The authors speculate that in patients with lacunar infarction “whether the atrial fibrillation was the cause of stroke, or merely a marker of coincidental atherosclerotic cardiac disease” (Sandercock et al. 1992). In general, clinical classification of ischemic strokes by presumed mechanism remains imperfect and inadequately validated (Hart et al. 2000). Conclusions drawn from incomplete data that lacks validation remain hypotheses.
Systemic embolism (SE) also occurs in patients with AF. AF may have other cumulative, as opposed to acute, damaging effects. AF may have a cumulative negative effect on the brain independent of cerebral infarcts; it may be associated with such problems as reduced brain volume and cognitive function independent of infarcts (Stefansdottir et al. 2013). There is also an association with silent cerebral infarctions, the significance of which is unclear (Kalantarian 2014). This circumstance is worrisome as there is much we do not know about the brain, particularly in regards to what happens over time.
Stroke risk prediction tools have been developed in patients with AF for risk stratification purposes. Many stroke risk factors commonly coexist with AF, such as hypertension, increasing age, diabetes mellitus, and other cardiovascular diseases. While these risk stratification schemes based on clinical factors such as CHADS2 and CHA2Ds2-VASC2 can help guide preventive therapy, these estimates have only weak or modest predictive value for identifying high risk patients (Lip and Lane 2015). CHA2DS2-VASC2 is best used to identify low risk patients who do not need antithrombotic therapy (Lip and Lane 2015). Due to low confidence in current risk stratification tools, better risk stratification tools need to be developed.
The mechanism for ischemic stroke and SE that is believed to be directly related to AF is thromboembolism. The pathogenesis of thromboembolism in AF is multifactorial. It includes Virchow’s triad of endothelial or endocardial damage or dysfunction, abnormal blood stasis and altered hemostasis, platelet function, and fibrinolysis (Watson et al. 2009). There is evidence for endothelial damage in the left atrium, particularly in the left atrial appendage which is a blind ended passage that has a narrow inlet, where there is particularly low flow. There is also evidence for a prothrombotic or hypercoaguable state where evidence has focused on the potential role of inflammation (Watson et al. 2009). “The mechanisms underlying thrombogenesis in atrial fibrillation are clearly complex and remain only partly understood” (Watson et al. 2009). As more precise knowledge is gained about AF and thromboembolism there may be increased methods of effective prevention.
Effective thromboprophylaxis is central to stroke prevention in patients with AF. The evidence is strong that anticoagulation with warfarin for patients with AF reduces fatal and nonfatal stroke by about 50%. This risk reduction is closely accompanied by an increased potential for bleeding as the factors for both are similar. “… the quality of anticoagulation control is critical (as reflected by the time in therapeutic range [TTR], with a target INR of 2.0-3.0). Time in therapeutic ranges are closely related to efficacy and safety, with low rates of stroke and bleeding with high TTRs” (Lip and Lane 2015). Several bleeding risk scores have been developed and validated in an AF population; in particular, the HAS-BLED score (Lip and Land 2015). Newer generation anticoagulants (NOACs) such as dabigatran, apixaban, rivaroxaban and edoxaban have proven efficacy with a reduced risk of hemorrhagic stroke and a potential for additional benefits. Current evidence for antiplatelet therapy (aspirin monotherapy and aspirin and clopidogrel dual therapy) is not straightforward; however, the potential for bleeding is present. The ACCP estimate benefits and risks of antithrombotic therapy in patients with AF (You et al. 2012):
- Antiplatelet monotherapy is associated with a 21% relative reduction in risk of nonfatal stroke (ischemic and hemorrhagic) compared with no treatment and a 50%-60% relative increase in the risk of major extracranial bleeding (pooled estimate).
- Dual antiplatelet therapy is associated with a 28% relative reduction in the risk of nonfatal stroke (ischemic and hemorrhagic) compared with antiplatelet monotherapy and a 50% relative increase in the risk of major extracranial bleeding (single trial).
Combining warfarin with antiplatelet therapy increases the risk for bleeding with no apparent benefit in thromboembolism reduction in patients with NVAF (Dentali et al. 2007).
The rationale for LAA occlusion is to exclude the LAA as a thromboembolic source. Echocardiographic and anatomic (autopsy and surgical) studies have demonstrated that when thrombus is observed in the left atrium, it is located in the LAA in >90% of cases (Whitlock et al. 2013)(Onalan and Crystal 2007)(Blackshear and Odell 1996).
Page 624. Table 1. Review of published reports detailing the frequency and site of thrombus location in patients with nonrheumatic atrial fibrillation. Onalan and Crystal. Left atrial appendage exclusion for stroke prevention in patients with nonrheumatic atrial fibrillation. Stroke 2007;38[part 2]: 624-630.
The LAA has a variable structure. To what degree certain aspects such as size and other characteristics influence thrombus formation is not completely known. Surgical exclusion incident to heart surgery has been done for decades; however, many of these patients remained on warfarin due to other indications such as valve replacement (Meier et al. 2014). There are potential concerns with LAA exclusion. Incomplete occlusion has been demonstrated in surgical cases, which may predispose to thrombus (Onalan and Crystal 2007). Percutaneous procedures and devices have been developed. Procedural and device-related risks include over or undersizing of device, device migration, dislodgement or embolization, cardiac perforation, and repeat procedures. There may be a persistent atrial septal defect secondary to atrial septal puncture. There is a potential for adjacent structure damage, including the left pulmonary veins and coronary arteries. Hemopericardium and resultant cardiac tamponade has occurred. The LAA also functions as a decompression chamber during left ventricular systole and during other periods when left atrial pressure is high, and functions as storage for atrial natriuretic factor, which performs an important physiologic function regulating the intravascular volume via release of atrial natriuretic peptide (Sakellaridis et al. 2014). “Therefore it is possible that the LAA maintains left atrial pressure not only through its increased distensibility and its capacity for ANF secretion, but also through stretch sensitive receptors which, when activated, increase heart rate, diuresis, and natriuresis” (Al-Saady et al. 1999). It is unclear what effect LAA occlusion has on these functions and if clinically significant adverse effects will result over the lifetime of the individual.
The WATCHMAN device
WATCHMAN is the only device that has been systematically studied with randomized controlled trial data and is the only FDA approved device in the U.S. specifically for LAA occlusion. The WATCHMAN device has two randomized controlled trials with Bayesian adaptive designs for FDA premarketing approval and an unprecedented three FDA advisory panel meetings prior to approval in March 2015. The first trial, PROTECT-AF, enrolled between February 2005 and June 2008. This trial met noninferiority based on prespecified stopping at the first interim analysis of 600 patient-years (pt-yrs). In August 2008, a PMA was filed with an analysis of the 600 pt-yr cohort. In November 2008, a major deficiency letter was issued with a response submitted by the sponsor in January 2009. In March 2009, a report containing an analysis of 900 patient-hours was provided to the FDA.
The first FDA advisory panel review was in April 2009. The advisory panel voted for approval with conditions but the FDA concluded that the study results did not demonstrate a reasonable assurance of device safety and effectiveness. A non-approvable letter was issued on March 11, 2010. The FDA worked with the sponsor to design a new clinical study, the PREVAIL trial. In July 2010, FDA conditionally approved the PREVAIL randomized controlled trial. In this Bayesian trial, interim analyses were designed to incorporate patients into an informative prior from the PROTECT AF trial with a 50% discount. The PROTECT AF patients are a subset that would have been eligible for the PREVAIL trial.
The second FDA advisory panel on December 11, 2013 reviewed the results of PREVAIL and additional long-term follow-up data from PROTECT AF (1588 pt-yrs and 2621 pt-yrs) and the Continued Access to PROTECT AF (CAP) Registry. The PREVAIL trial did not meet statistical noninferiority. The advisory panel voted for approval; however in February 2014 the FDA received updated follow-up data from the PREVAIL trial that showed additional ischemic strokes in WATCHMAN device subjects, calling into question the mechanism of the device.
The third advisory panel meeting to discuss updated datasets from the PREVAIL and PROTECT AF trials was in October 2014. Ultimately, the WATCHMAN device received FDA approval in March 2015.
The first PMA trial, PROTECT AF, had the following major issues that create low confidence in the ability to determine patient benefit.
- Patient population.
- The trial was designed to demonstrate the safety and efficacy of the WATCHMAN device in patients with NVAF and a CHADS2 of 1 or more. This patient population was weighted toward lower risk patients based on the CHADS2 score: 68% CHADS2 1 or 2 versus 5% CHADS2 5 or 6 in the device arm. Further, patients with CHADS2 =1 (31%) may have been acceptable candidates for aspirin therapy rather than warfarin. Only about one-fifth of patients were at highest risk – those with a previous TIA or stroke where annual risk is about 10%-12%, too small a subpopulation to draw any conclusions from.
- It would seem that the device would have the greatest benefit in those ineligible for anticoagulants, however a patient requirement was that they had to be eligible for long term warfarin therapy.
- The list of trial eligibility criteria had 30+ inclusion/exclusions. Of all patients screened, 86% were deemed ineligible, calling into question the applicability of results to real world patients in seen in clinic.
- Confounding by medical therapies given and not given in both the device arm and the control arm, creating difficulty in interpretation of events between groups.
- The effect of confounding by warfarin continuation in device patients and failure to take warfarin or discontinuation in the control patients creates a situation where you cannot separate treatment effects. An attempt to separate effects with the sponsor PP analysis has limitations in that it biased the results to favor the device.
- Concomitant use of chronic clopidogrel therapy in both study groups with imbalance (51% of follow-up time in device subjects and 16% of follow-up time in control subjects) adds to a lack of clarity. Similarly for aspirin therapy, the percentage of follow-up duration that patients in the device group were 91% versus 54% in the control group. The antiplatelet drugs could act as protection for ischemic events or may contribute to bleeding, including hemorrhagic stroke. Of the 6 control patients with hemorrhagic stroke, 2 were on aspirin and warfarin, one was on aspirin, clopidogrel and warfarin, and the antiplatelet drug status of the remaining three is not known. Combining warfarin with antiplatelet therapy increased the risk for bleeding with no apparent benefit in thromboembolism reduction in patients with NVAF (Dentali et al. 2007).
Note in PROTECT AF the ischemic stroke rate favored the control group (3.0% in the device group versus 2.0% in the warfarin group). In the device group 1 stroke was before the procedure and 5 were procedural events. The primary endpoint also included hemorrhagic stroke. There were six hemorrhagic strokes in the warfarin group corresponding to a higher rate than observed in contemporary anticoagulation clinical trials.
Safety of the procedure as reflected in a high serious peri-procedural event rate was a major issue in the first FDA advisory panel review. The procedure is technically demanding requiring skill at transseptal puncture and manipulation of the device into a thin LAA. The procedure can result in major adverse events including device embolization, air embolus, pericardial effusions and death. Work by the sponsor to improve safety is reflected in subsequent publications including results in the FDA Executive Summaries. Operators followed the sponsor recommended qualifications and training, taking into account the level of investigator qualifications, and safety of the procedure improved. Device implantation by new operators was not associated with reduced rates of implant success or an increased risk of major adverse events.
The PREVAIL trial was similar in design to PROTECT AF with the noted exception of the PROTECT AF limitations that PREVAILwas designed to address: (FDA Executive Summary 2013):
- PREVAIL enrolled higher risk patients than PROTECT AF.
- PREVAIL excluded patients indicated for chronic clopidogrel therapy to avoid this confounding issue.
- The sponsor committed to improved enhanced monitoring of warfarin use to ensure adequate compliance and improved INR control.
- The noninferiority margin for the event rate ratio for the primary effectiveness endpoint was reduced to 1.75.
- The second primary endpoint was developed to compare the rate of ischemic stroke and SE beyond the first 7 days post-randomization to exclude peri-procedural events, a hypothesized “proof of concept” endpoint as the device is supposed to prevent thromboembolic events.
- A third primary endpoint to reflect acute device safety comprised of defined events occurring between the time of randomization and within 7 days of the procedure or by hospital discharge.
- An evaluation of the procedural learning curve.
Similar to PROTECT AF, the PREVAIL study enrolled predominately male (about 70%) and Caucasian (about 94%) patients. There was a statistically significant difference in the percentage of patients with hypertension (88.5% in the device group versus 97.1% in the control group, p = 0.003), the most important risk factor for stroke. Data from PROTECT AF and the CAP registry was used as prior information in the Bayesian statistical analysis of the primary endpoints.
PREVAIL results at the second FDA panel meeting:
- The first primary effectiveness endpoint based on PREVAIL (only 28% of subjects had 18 months of follow-up) and historical PROTECT AF data did not meet noninferiority.
- The second primary endpoint had two noninferiority criteria, if either was met the endpoint would be satisfied. Noninferiority was narrowly met for only one criterion. As with the first primary endpoint, only 28% of PREVAIL subjects had 18 months of data. However, in PREVAIL, the event rates favored the control group.
- Success for the third primary endpoint was based on a prespecifed performance goal and was intended to reflect procedural safety. This endpoint was met.
- Though not designed to compare major bleeding rates between arms, there did not appear to be a signal of a reduction in total bleeding events or a reduction in bleeding events over time in the WATCHMAN device group compare to the warfarin arm.
- Thirteen case of device thrombus (not included as serious adverse events).
Longer-term follow-up data from PROTECT AF examined at the second FDA advisory panel is noted for the following:
- “The rate of subject withdrawal, particularly the disparity in withdrawal rates between treatment groups, could lead to bias against the control group and favoring of the device group for the long-term event rate comparisons presented below, considering that the hazard rates decrease over time.”
- The primary effectiveness endpoint was met for statistical noninferiority. However, “…the model used to estimate the event rates is not accurate and sole reliance on the statistical results from the primary analysis may be problematic.”
- Device implantation is associated with a higher rate of safety events as compared with medical therapy.
- PROTECT AF was not designed to detect a difference in major bleeding rates, though the numerical trend in this trial appears to favor the device.
PREVAIL results at the third FDA panel meeting:
- PREVAIL data was no longer dominated by prior data.
- A further divergence in the rate of ischemic stroke and SE, significantly favoring the control group. There appeared to be no mitigating circumstances that could otherwise address device effectiveness concerns.
- Hemorrhagic stokes were infrequent.
- The hemorrhagic stroke rate in the warfarin group was in the range observed in the warfarin groups of contemporary anticoagulation trials.
- The rate of cardiovascular or unexplained death numerically favored the device group but none of the deaths was causally linked to the WATCHMAN device,
implantation procedure, or anticoagulant therapy.
- The first primary endpoint was more distant from the noninferiority success criterion.
- The second primary endpoint no longer met noninferiority success criterion.
- The PREVAIL data are noted to be divergent from the PROTECT AF data and further favor the warfarin group.
- Ten of 13 ischemic stroke events occurred more than one year after device implantation, calling into question the effectiveness of the device mechanism of action.
- The ischemic stroke plus SE rate in the PREVAIL CAP2 registry is generally similar to the rate seen in the PREVAIL trial.
Longer-term follow-up data from PROTECT AF examined at the third FDA advisory panel is noted for the following:
- The ischemic stroke plus SE rate numerically favored the warfarin group. The ischemic stroke and SE rate in the PROTECT AF CAP registry was generally similar.
- Results of the PROTECT AF suggest that the WATCHMAN device offers a benefit by lowering the risk of hemorrhagic stroke. However,
- The hemorrhagic stroke rate in the warfarin group was considerably higher than expected based on results of contemporary anticoagulation trials.
- A review of the clinical narratives of the 10 hemorrhagic strokes in the warfarin group reveals low confidence in reported difference due to non-use of warfarin in a subject, absence of imaging confirmation in one subject, concomitant use of antiplatelet agents in several subjects that potentially increased the bleeding risk, and events associated with trauma in several subjects (where subdural classification may have been more appropriate).
To summarize, the ischemic stroke plus SE rate favored the warfarin group in both the PROTECT AF and PREVAIL trials, with a significant difference in rates observed in the PREVAIL trial. The occurrence of the late ischemic strokes is particularly concerning and could be related to thrombus formation on the device in the absence of anticoagulation. The hemorrhagic stroke benefit of the device seen in PROTECT AF is of low confidence and a benefit was not seen in PREVAIL. Neither trial was designed to compare major bleeding rates. Although late bleeding favors the device, this is balanced by the procedure related bleeding, resulting in no overall device advantage. In PROTECT AF, the difference in cardiovascular and unexplained death rates is largely due to deaths related to hemorrhagic stroke, and in PREVAIL the device is also favored, but none of the deaths was causally linked to the device, procedure, or anticoagulant therapy. Any inference of all-cause mortality differences in PREVAIL are limited by subject comorbidities, where death did not appear to be associated with the device or anticoagulation.
The interpretation of evidence has these additional considerations:
- The demonstration of stroke prevention effectiveness should be robust when the risk of the intervention is high, or when there is a proven therapy, particularly so for patients who are likely not to experience the event that the intervention endeavors to prevent. When patient risk for the event or effectiveness is unclear, patients may be placed at risk without benefit.
- LAAC is intended to prevent thromboembolism for the LAA in patients with NVAF. However, the endpoint included hemorrhagic stroke and cardiovascular and unexplained death. The composite effectiveness endpoint events were events that would also be considered significant safety events - stroke, death, and systemic embolism. The number of these types of events would be expected to be low, particularly in a population with predominance of lower risk patients, thus lending itself to composite endpoint design to shorten the length of the trial. However, composite endpoint design creates difficulty in interpretation, particularly when the concepts of effectiveness, prevention in the case of the device and warfarin, and safety are co-mingled. Components of the composite endpoint should be examined to determine patient benefit.
- The effectiveness endpoint was designed for noninferiority, not equivalence. This meant that the stroke, death, and SE rate in the patients with the device could be two times, or in the case of the PREVAIL trial 1.75 times, the event rate in the warfarin group and still be considered a successful trial in favor of the device. Noninferiority trial designs can be used when there is an advantage of the experimental therapy. The advantage in this circumstance is hypothesized to be decreased major bleeding, including hemorrhagic stroke. The current data does not support a major bleeding advantage for the device.
- The clinical question is does this device without long-term warfarin provide equivalent protection against thromboembolic events to the AF patients compared to best medical therapy.
- Some guidelines now recommend NOAC consideration before warfarin. Each of the NOACs has a different benefit risk profile in comparison to warfarin in large RCTs. A reduction in hemorrhagic stroke and/or major bleeding in comparison to warfarin has been observed.
- Quality of OAC management is important and requires optimization. Improved quality of warfarin administration (in addition to better defined antiplatelet management) may partially explain the lower bleeding rate observed in the PREVAIL trial. In the PROTECT AF trial, all 5 of the ischemic strokes in the control group occurred either when the INR was subtherapeutic or when there was no data on INR within 30 days of the event; of the 6 control patients with hemorrhagic stroke, only 2 had INRs in the therapeutic range recoded within 30 days of the event. There were 40% of the control patients who had at least one INR > 4.0 during the study, while 26% of the WATCHMAN patients did. Changes in trial conduct and execution appear to affect the trial results.
In the confines of a clinical trial, in patients who were eligible for warfarin, the WATCHMAN patients had more ischemic strokes than the warfarin patients. These trials do not support a conclusion of reasonable and necessary for prevention of ischemic stroke in the Medicare population who is eligible for prophylaxis with warfarin.
Other additional clinically important questions remain with no current answer:
- Will device deployment, with the potential for opening up a right-to-left inter-atrial shunt, affect the long-term risk of paradoxical embolism, or alter the risk of cardiac arrhythmias and subsequent cardioembolic events and increase stroke rate in the Medicare population (McCabe et al. 2009)?
- What physiologic effects will the occlusion have on overall cardiac function and intravascular volume, particularly as patients age and cardiac function is reduced, and in the many Medicare patients who develop heart failure?
- What causes thrombus to form on the device and what is the time course of these events? LAA thrombus is the purported mechanism for stroke. It would seem that thrombus on the device would be placing patients at high risk for stroke.
- Other percutaneous procedures produce procedure-related emboli. What is procedural embolic potential with LAAC and what is the long-term clinical outcome?
- How does the device compare to what now is considered by some to be the first consideration for antithrombotic therapy, the NOACs?
The meta-analysis of WATCHMAN data adds little to the previous evidence base. Without a method to accurately adjudicate stroke etiology, it is possible that many of these strokes were unrelated to LAA thrombi (Waks and Manning 2015).
Studies without OACs
The WATCHMAN device may be best for those at high risk of thromboembolic stroke and who cannot take long term OACs or those who had events on OACs. However, the safety of the WATCHMAN device in patients who are poor candidates for OACs or antiplatelet therapy is unknown. A small nonrandomized trial did demonstrate reasonable safety over a short term. The Medicare population likely has a high percentage of these patients that could potentially have a significant health benefit, i.e., avoidance of stroke. Information concerning this therapy in these patients is of great interest.
Other Devices
Other catheter-based devices have been developed but only the WATCHMAN device has been systematically studied in randomized controlled trials and is FDA approved for this indication. The PLAATO device has several observational human studies. It used only antiplatelet therapy after implantation. This device was removed from the market in 2006 and was not approved for use in the United States. The Amplatzer devices reportedly have the longest clinical follow-up of currently available LAA occluders. However, this device has only low quality observational studies without a comparator and is not FDA approved for this purpose. The LARIAT technique has only very preliminary data. The off-label use of these devices has minimal evidentiary support.
These devices with various designs/mechanisms of action that are either not FDA approved or are used off-label for this indication do not have adequate data to demonstrate a health benefit and could have the potential for harm. For example, one device has a recent safety warning (http://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm454501.htm).
Guidelines and Consensus for percutaneous LAA closure for stroke prevention in AF
The American Heart Association/American Stroke Association makes this recommendation:
Closure of the LAA may be considered for high-risk patients with AF who are deemed unsuitable for anticoagulation if performed at a center with low rates of periprocedural complications and the patient can tolerate the risk of at least 45 days of postprocedural anticoagulation (Class IIb; Level of Evidence B).
The American College of Chest Physicians and the American Academy of Neurology make no formal recommendation for percutaneous LAA closure for stroke prevention.
The Canadian Cardiovascular Society does not recommend use except in research protocols for those at high risk who are unable to take anticoagulants. The British National Health Service Commissioning Board ruled that the cost-effectiveness and clinical effectiveness of the device are not established. The consensus statement by the European Heart Rhythm Association and the European Association of Percutaneous Cardiovascular Interventions (EAPCI) includes, “Patients with a high thrombo-embolic risk (CHA2DS2-VASc score of > 2) but contraindication to oral and systemic anticoagulation (e.g. history of a significant bleeding event such as intracranial or life-threatening bleeding, the source of which cannot be eliminated) represent the most accepted clinical indication for LAA occlusion, albeit by having to extrapolate the results of the PROTECT AF study to that specific cohort.” As an alternative to oral anticoagulation when oral anticoagulant is possible, they recommend the mention of LAA occlusion as in Figure 5 of the Consensus Statement.
Guidelines and consensus support further study of LAA occlusion. Coverage with evidence development supports attainment of this goal and has criteria that support patient safety, transparency, and dissemination of information. Investigators must register at ClinicalTrials.gov. Registrants at ClinicalTrials.gov must submit a standardized set of data elements to describe the study design, eligible populations, outcome measures, and other parameters and results. Registration on this site, for most studies, serves as a vehicle for Medicare beneficiaries to learn about, and identify studies in which they may want to participate. When reporting of results are required, it also offers an assurance of quality because, generally, public access to information enables a higher level of accountability in the accurate reporting of the clinical study protocol and results, and in the conduct of the trial itself. This accountability derives both from public access to information about studies and from the risk of penalty for submitting false or misleading clinical trial information. Registration with ClinicalTrials.gov also assures that Medicare beneficiaries and their treating healthcare professionals will have pertinent information about CED studies, and we expect this may facilitate better informed decision-making. Similarly, registry studies that registered at AHRQ’s RoPR are advised to follow the set of best practices on methodologies and on the technical, legal, ethical, and analytical considerations for designing, operating, and utilizing registries and registry data as described in AHRQ’s Registries for Evaluating Patient Outcomes: A User’s Guide.
Current limitations of warfarin therapy
Despite warfarin being effective in stroke prevention, warfarin discontinuation rates may be as high as 38% a year, and about 14-44% of at-risk patients are said to be ineligible for treatment (Wrigley and Lip 2009) (Gomes et al. 2012). Warfarin is underutilized for a variety of reasons including risks of major bleeding and falls, inconvenience of frequent monitoring and dose adjustments, drug interactions, and restrictions on diet and alcohol intake. It is particularly underutilized in the elderly, who are at greatest risk. Provider initiated therapy discontinuation may also play a role, rather than patient non-adherence or adverse events (Maxwell and Bennett 2012). These circumstances highlight that provider-patient shared decision making is important.
Shared Decision-Making (SDM)
Patient shared decision-making is especially important in treatments where there are complex considerations on benefits, harms, indications and existing equally effective treatments. Stroke prophylaxis with antithrombotic therapy is recognized as having complex considerations. A systematic review on patient values and preferences in decision making for antithrombotic therapy found that values and preferences were highly variable among individuals (MacLean et al. 2012). The importance of individual patient values and preferences in decision making applies to LAAC therapy. The shared decision making interaction will assist in properly selecting patients for LAAC (86% of patients did not meet the specific study criteria of pivotal trials). An evidence-based decision support tool on anticoagulation in patients with NVAF must be used to help patients better understand treatment options and choose the most appropriate treatment course. SDM must occur prior to LAAC, must be documented in the medical records, must include a discussion of the benefits and harms, must document an appropriate rationale to seek a non-pharmacologic alternative to warfarin, taking into account the safety and effectiveness of the device compared to anticoagulation, and result, after being informed of the reported risks of LAAC and reasonable alternative management strategies, in adequately informed consent.
Health Disparities
Gender and racial disparities in many trials continue. At face value, trial demographics should reflect the prevalence in the population that would be eligible for treatment, regardless of age, gender, race, or socioeconomic status. In PROTECT AF there was a disproportionate number of male patients (70%) compared to female patients (30%) as well as a disproportionate number of Caucasian patients (91%).
Disparities in AF related stroke and mortality persist. Barriers to treatment are found to include lack of consistency in assessing stroke and anticoagulant-related bleeding risk, underuse of standardized risk assessment tools, discomfort with novel anticoagulants, and patient education deficiencies (Karcher et al. 2015). Furthermore, overall disparities in cardiovascular care persist.
Summary
Atrial fibrillation management is an area of rapid changes. There is still a surprising amount of information that we either do not know or cannot be confident of. In review of the totality of the percutaneous LAA closure data, with consideration of each component of the evidence, a risk benefit profile for LAA closure cannot be determined. This is particularly true in consideration for an individual patient to determine his or her own risk benefit profile. The LAA information for decision making is either unclear or unavailable. There is a need for improved stroke and bleeding risk stratification and subsequent accurate communication tools to enable improved individualized decision making. There is a need to understand the use of antiplatelet therapy after device implantation as opposed to anticoagulant use. There is a need to understand the performance of this device in comparison to NOACs. Appropriate use of anticoagulants is underutilized, particularly among the elderly who have the highest prevalence of AF. There is an NVAF population with high CHADS2 or CHA2DS2-VASc Scores, a high HAS-BLED score and a clearly defined contraindication to warfarin. These patients, many of whom are in the Medicare population, would be most likely to benefit from this type of therapy. While CMS has embraced an evidence-based medicine coverage paradigm, CMS is increasingly challenged to respond to requests for coverage of certain items and services when we find that the expectations of interested parties are disproportionate to the existing evidence base. At the same time, we believe that CMS should support evidence development for certain innovative technologies that are likely to show benefit for the Medicare population, but where the available evidence base does not provide a sufficiently persuasive basis for coverage outside the context of a clinical study, which may be the case for new technologies, or for existing technologies for which the evidence is incomplete.
Coverage in the context of ongoing clinical research protocols or with additional data collection can expedite earlier beneficiary access to innovative technology while ensuring that systematic patient safeguards, including assurance that the technology is provided to clinically appropriate patients, are in place to reduce the risks inherent to new technologies, or to new applications of older technologies.
At this time, there is insufficient evidence to determine that percutaneous LAA closure using an implanted device improves health outcomes compared to non-invasive standard medical therapies for Medicare beneficiaries with NVAF. Therefore, LAA closure using an implanted device should only be considered in patients with NVAF with a high stroke risk and a contraindication to anticoagulation (e.g. history of a significant bleeding event such as intracranial or life-threatening bleeding, the source of which cannot be eliminated) in a clinical trial or comparative study.
IX. Conclusion
A. The Centers for Medicare & Medicaid Services (CMS) proposes that the evidence is sufficient to determine percutaneous left atrial appendage closure (LAAC) therapy using an implanted device is not reasonable and necessary to diagnose or treat an illness or injury or to improve the functioning of a malformed body member and, therefore, is not covered under § 1862(a)(1)(A) of the Social Security Act.
B. In order to support evidence development for technologies likely to show benefit for the Medicare population, CMS proposes to cover items or services that are reasonable and necessary for research under §1862(a)(1)(E) of the Social Security Act using the Coverage with Evidence Development (CED) paradigm. We propose that coverage would be limited to items and services in clinical studies that meet the conditions specified below:
Percutaneous LAAC therapy is covered for patients with non-valvular atrial fibrillation only, when all of the following conditions 1-7 are met.
- The device is FDA approved for patients with non-valvular atrial fibrillation.
- The patient has:
- A high CHADS2 (Congestive heart failure, Hypertension, Age >75, Diabetes, Stroke/transient ischemia attack/thromboembolism) or CHA2DS2-VASc score (Congestive heart failure, Hypertension, Age ≥ 65, Diabetes, Stroke/transient ischemia attack/thromboembolism, Vascular disease, Sex category); and
- A high HAS-BLED score (Hypertension, Abnormal renal function and/or liver function, Stroke, prior Bleeding, Labile anticoagulation range, Elderly age>65, Drug therapy such as antiplatelet drugs), and
- A contraindication to warfarin.
- The procedure is furnished in a hospital that meets the following institutional requirements:
- Cardiac catheterization lab or electrophysiology (EP) lab with fluoroscopy capability
- Non-invasive imaging (i.e. transesophageal echocardiography) with dedicated echocardiography support
- Anesthesiology support for administration of general anesthesia specific to this procedure
- Cath lab, operating room (if required), post anesthesia recovery, intensive care and step down unit space to accommodate cases with and without complications
- On-site emergency cardiac surgery services
- The procedure is performed by physicians with the following qualifications and experience:
- The primary implanting physician has performed ≥ 25 interventional cardiac procedures involving a trans-septal puncture (TSP) in their total experience, with at least 10 TSP procedures performed over the past 12 month period.
- The primary implanting physician(s) must be an interventional cardiologist and/or an electrophysiologist. They may jointly participate in intra-procedural aspects of the implant or perform the implant procedure individually.
- The interventional cardiologist(s) and electrophysiologist(s) must receive the training prescribed by the manufacturer on the safe and effective use of the device prior to performing implant procedures. Training should also include a physician who already has experience implanting the device, but who is not a representative of the manufacturer, as well as a minimum of two supervised and two observed cases.
- The patient is enrolled in, and the treating physician team is participating in a prospective national registry that consecutively enrolls LAAC patients and tracks the following annual outcomes at the patient data level for a period of at least five years from the time of the LAAC procedure.
- Operator-specific complications
- Device-specific complications including device thromboses
- Stroke, adjudicated, by type
- TIA
- Systemic embolism
- Death
- Major bleeding, by site and severity
The registry must be designed to permit identification and analysis of patient, practitioner and facility level factors that predict patient risk for these outcomes. The registry must include contemporaneous patients followed on oral anticoagulant (OAC) therapy to serve as non-interventional controls. CMS will review the qualifications of candidate registries to ensure that the approved registry follows standard data collection practices and collects data necessary to evaluate the patient outcomes specified above.
The registry must collect all data necessary to conduct multivariable adjusted analyses and have a written executable analysis plan in place to address the following questions:
In a prospective, clinical study, when percutaneous LAAC therapy using an implanted device is performed outside a randomized, controlled clinical trial, compared to non-interventional controls followed on oral anticoagulant (OAC) therapy:
- How do the outcomes listed above compare to outcomes in the pivotal clinical trials in the short term (<12 months) and in the long term (≥ 5 years)?
- How do the outcomes listed above differ from outcomes in comparable concurrent OAC controls in the short term (<12 months) and in the long term (>5 years)?
- What is the long term (≥ 5 year) durability of the device?
- What are the short term (<12 months) and the long term (>5 years) device-specific complications including device thromboses?
- A formal shared decision-making interaction between the patient and provider using an evidence-based decision tool on anticoagulation in patients with NVAF must occur prior to LAAC, must be documented in the medical records, must include a discussion of the benefits and harms, must document an appropriate rationale to seek a non-pharmacologic alternative to anticoagulants, taking into account the safety and effectiveness of the device compared to anticoagulants, and have, after being informed of the reported risks of LAAC and reasonable alternative management strategies, given informed consent.
All CMS-approved clinical studies and registries must adhere to the following standards of scientific integrity and relevance to the Medicare population:
- The principal purpose of the study is to test whether the item or service meaningfully improves health outcomes of affected beneficiaries who are represented by the enrolled subjects.
- The rationale for the study is well supported by available scientific and medical evidence.
- The study results are not anticipated to unjustifiably duplicate existing knowledge.
- The study design is methodologically appropriate and the anticipated number of enrolled subjects is sufficient to answer the research question(s) being asked in the National Coverage Determination.
- The study is sponsored by an organization or individual capable of completing it successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found in the Code of Federal Regulations (CFR) at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it is also in compliance with 21 CFR Parts 50 and 56. In addition, to further enhance the protection of human subjects in studies conducted under CED, the study must provide and obtain meaningful informed consent from patients regarding the risks associated with the study items and/or services, and the use and eventual disposition of the collected data.
- All aspects of the study are conducted according to appropriate standards of scientific integrity.
- The study has a written protocol that clearly demonstrates adherence to the standards listed here as Medicare requirements.
- The study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Such studies may meet this requirement only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research studies and registries are registered on the www.ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject. Registries are also registered in the Agency for Healthcare Quality (AHRQ) Registry of Patient Registries (RoPR).
- The research study protocol specifies the method and timing of public release of all prespecified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 12 months of the study’s primary completion date, which is the date the final subject had final data collection for the primary endpoint, even if the trial does not achieve its primary aim. The results must include number started/completed, summary results for primary and secondary outcome measures, statistical analyses, and adverse events. Final results must be reported in a publicly accessibly manner; either in a peer-reviewed scientific journal (in print or on-line), in an on-line publicly accessible registry dedicated to the dissemination of clinical trial information such as ClinicalTrials.gov, or in journals willing to publish in abbreviated format (e.g., for studies with negative or incomplete results).
- The study protocol must explicitly discuss beneficiary subpopulations affected by the item or service under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations in the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The study protocol explicitly discusses how the results are or are not expected to be generalizable to affected beneficiary subpopulations. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions. All other indications are nationally non-covered.
The principal investigator must submit the complete study protocol, identify the relevant CMS research question(s) that will be addressed and cite the location of the detailed analysis plan for those questions in the protocol, plus provide a statement addressing how the study satisfies each of the standards of scientific integrity (a. through m. listed above), as well as the investigator’s contact information, to the address below. The information will be reviewed, and approved studies will be identified on the CMS website.
Director, Coverage and Analysis Group
Re: LAAC Therapy CED
Centers for Medicare & Medicaid Services (CMS)
7500 Security Blvd., Mail Stop S3-02-01
Baltimore, MD 21244-1850
We are requesting public comments on this proposed determination pursuant to section 1862(l) of the Social Security Act. We are specifically interested in public comments on the use of Coverage with Evidence Development (CED) in this decision. After considering the public comments, we will make a final determination and issue a final decision memorandum.
APPENDIX A
General Methodological Principles of Study Design
(Section VI of the Decision Memorandum)
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of an illness or injury or to improve the functioning of a malformed body member. The overall objective for the critical appraisal of the evidence is to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for patients.
We divide the assessment of clinical evidence into three stages: 1) the quality of the individual studies; 2) the generalizability of findings from individual studies to the Medicare population; and 3) overarching conclusions that can be drawn from the body of the evidence on the direction and magnitude of the intervention’s potential risks and benefits.
The methodological principles described below represent a broad discussion of the issues we consider when reviewing clinical evidence. However, it should be noted that each coverage determination has its unique methodological aspects.
Assessing Individual Studies
Methodologists have developed criteria to determine weaknesses and strengths of clinical research. Strength of evidence generally refers to: 1) the scientific validity underlying study findings regarding causal relationships between health care interventions and health outcomes; and 2) the reduction of bias. In general, some of the methodological attributes associated with stronger evidence include those listed below:
- Use of randomization (allocation of patients to either intervention or control group) in order to minimize bias.
- Use of contemporaneous control groups (rather than historical controls) in order to ensure comparability between the intervention and control groups.
- Prospective (rather than retrospective) studies to ensure a more thorough and systematical assessment of factors related to outcomes.
- Larger sample sizes in studies to help ensure adequate numbers of patients are enrolled to demonstrate both statistically significant as well as clinically significant outcomes that can be extrapolated to the Medicare population. Sample size should be large enough to make chance an unlikely explanation for what was found.
- Masking (blinding) to ensure patients and investigators do not know to which group patients were assigned (intervention or control). This is important especially in subjective outcomes, such as pain or quality of life, where enthusiasm and psychological factors may lead to an improved perceived outcome by either the patient or assessor.
Regardless of whether the design of a study is a randomized controlled trial, a non-randomized controlled trial, a cohort study or a case-control study, the primary criterion for methodological strength or quality is the extent to which differences between intervention and control groups can be attributed to the intervention studied. This is known as internal validity. Various types of bias can undermine internal validity. These include:
- Different characteristics between patients participating and those theoretically eligible for study but not participating (selection bias).
- Co-interventions or provision of care apart from the intervention under evaluation (performance bias).
- Differential assessment of outcome (detection bias).
- Occurrence and reporting of patients who do not complete the study (attrition bias).
In principle, rankings of research design have been based on the ability of each study design category to minimize these biases. A randomized controlled trial minimizes systematic bias (in theory) by selecting a sample of participants from a particular population and allocating them randomly to the intervention and control groups. Thus, in general, randomized controlled studies have been typically assigned the greatest strength, followed by non-randomized clinical trials and controlled observational studies. The design, conduct and analysis of trials are important factors as well. For example, a well designed and conducted observational study with a large sample size may provide stronger evidence than a poorly designed and conducted randomized controlled trial with a small sample size. The following is a representative list of study designs (some of which have alternative names) ranked from most to least methodologically rigorous in their potential ability to minimize systematic bias:
- Randomized controlled trials
- Non-randomized controlled trials
- Prospective cohort studies
- Retrospective case control studies
- Cross-sectional studies
- Surveillance studies (e.g., using registries or surveys)
- Consecutive case series
- Single case reports
When there are merely associations but not causal relationships between a study’s variables and outcomes, it is important not to draw causal inferences. Confounding refers to independent variables that systematically vary with the causal variable. This distorts measurement of the outcome of interest because its effect size is mixed with the effects of other extraneous factors. For observational, and in some cases randomized controlled trials, the method in which confounding factors are handled (either through stratification or appropriate statistical modeling) are of particular concern. For example, in order to interpret and generalize conclusions to our population of Medicare patients, it may be necessary for studies to match or stratify their intervention and control groups by patient age or co-morbidities.
Methodological strength is, therefore, a multidimensional concept that relates to the design, implementation and analysis of a clinical study. In addition, thorough documentation of the conduct of the research, particularly study selection criteria, rate of attrition and process for data collection, is essential for CMS to adequately assess and consider the evidence.
Generalizability of Clinical Evidence to the Medicare Population
The applicability of the results of a study to other populations, settings, treatment regimens and outcomes assessed is known as external validity. Even well-designed and well-conducted trials may not supply the evidence needed if the results of a study are not applicable to the Medicare population. Evidence that provides accurate information about a population or setting not well represented in the Medicare program would be considered but would suffer from limited generalizability.
The extent to which the results of a trial are applicable to other circumstances is often a matter of judgment that depends on specific study characteristics, primarily the patient population studied (age, sex, severity of disease and presence of co-morbidities) and the care setting (primary to tertiary level of care, as well as the experience and specialization of the care provider). Additional relevant variables are treatment regimens (dosage, timing and route of administration), co-interventions or concomitant therapies, and type of outcome and length of follow-up.
The level of care and the experience of the providers in the study are other crucial elements in assessing a study’s external validity. Trial participants in an academic medical center may receive more or different attention than is typically available in non-tertiary settings. For example, an investigator’s lengthy and detailed explanations of the potential benefits of the intervention and/or the use of new equipment provided to the academic center by the study sponsor may raise doubts about the applicability of study findings to community practice.
Given the evidence available in the research literature, some degree of generalization about an intervention’s potential benefits and harms is invariably required in making coverage determinations for the Medicare population. Conditions that assist us in making reasonable generalizations are biologic plausibility, similarities between the populations studied and Medicare patients (age, sex, ethnicity and clinical presentation) and similarities of the intervention studied to those that would be routinely available in community practice.
A study’s selected outcomes are an important consideration in generalizing available clinical evidence to Medicare coverage determinations. One of the goals of our determination process is to assess health outcomes. We are interested in the results of changed patient management not just altered management. These outcomes include resultant risks and benefits such as increased or decreased morbidity and mortality. In order to make this determination, it is often necessary to evaluate whether the strength of the evidence is adequate to draw conclusions about the direction and magnitude of each individual outcome relevant to the intervention under study. In addition, it is important that an intervention’s benefits are clinically significant and durable, rather than marginal or short-lived. Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits.
If key health outcomes have not been studied or the direction of clinical effect is inconclusive, we may also evaluate the strength and adequacy of indirect evidence linking intermediate or surrogate outcomes to our outcomes of interest.
Assessing the Relative Magnitude of Risks and Benefits
Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits. Health outcomes are one of several considerations in determining whether an item or service is reasonable and necessary. For most determinations, CMS evaluates whether reported benefits translate into improved health outcomes. CMS places greater emphasis on health outcomes actually experienced by patients, such as quality of life, functional status, duration of disability, morbidity and mortality, and less emphasis on outcomes that patients do not directly experience, such as intermediate outcomes, surrogate outcomes, and laboratory or radiographic responses. The direction, magnitude and consistency of the risks and benefits across studies are also important considerations. Based on the analysis of the strength of the evidence, CMS assesses the relative magnitude of an intervention or technology’s benefits and risk of harm to Medicare beneficiaries.
[1] Class IIb: Benefit ≥ risk; additional studies with broad objectives needed; additional registry data would be helpful. Procedure/treatment may be considered.
Level of Evidence B: Limited populations evaluated. Data derived from a single randomized trial or nonrandomized studies.


