To: Administrative File: CAG # 00413N
Erythropoiesis Stimulating Agents (ESAs) for Treatment of Anemia in Adults with CKD
Including Patients on Dialysis and Patients not on Dialysis
From: Louis B. Jacques, MD
Director, Coverage and Analysis Group
Tamara Syrek Jensen, JD
Deputy Director, Coverage and Analysis Group
James Rollins, MD, PhD
Division Director
Kimberly Long
Lead Analyst
Elizabeth Koller, MD
Lead Medical Officer
Subject: Final Decision Memorandum for CAG # 00413N
Erythropoiesis Stimulating Agents (ESA) for Treatment of Anemia in Adults with CKD
Including Patients on Dialysis and Patients not on Dialysis
Date: June 16, 2011
I. Final Decision
Given the totality of the currently available evidence, CMS will not issue a national coverage determination at this time for Erythropoiesis Stimulating Agents (ESAs) for Treatment of Anemia in Adults with CKD Including Patients on Dialysis and Patients not on Dialysis (CAG-00413N).
II. Background
In this section, we describe the technological developments that gave rise to recombinant erythropoietin and related erythrocyte stimulating agents (ESAs). We then describe the physiologic role of the kidneys, pathology of renal disease, and the demographics of renal disease. This is followed by a description of the types of anemia found in renal disease. Finally, we describe how anemia management has changed over time. For purposes of this discussion, therapy for a medical condition includes treatment for the signs and symptoms of the underlying condition. Though we have tried to simplify the discussion for the lay reader, the topic is scientifically complex and we believe that an overly simplistic treatment would ultimately be detrimental to the understanding of our review. We caution the reader that the term “inulin” refers to a polysaccharide used to measure kidney function and should not be misread as the term “insulin.”
ERYTHROPOIETIN IN RENAL DISEASE
A. Biochemical Background
Erythropoietin is a 34-kDa glycoprotein hormone produced primarily, but not exclusively, in the kidney and to a lesser extent in the liver. (Miyake 1977) The native protein is a 193 amino acid peptide sequence from which a 27 amino acid peptide leader sequence is removed from the N-terminus. An arginyl residue at the carboxyl terminus also appears to be cleaved during post-translation processing. The mature protein consists of a 165 amino acid backbone with 2 disulfide bonds, three N-linked carbohydrate chains, and one O-linked carbohydrate chain. The major side chains, sialated tetra-antennary saccharides, contribute to in vivo stability (Faults 1989).
As indicated above, production of this hormone is controlled via a feedback loop. (Bauer 1898, Erslev 1980)) Anemia and/or hypoxia result in decreased oxygen tension at the tissue level. Via intermediate signaling, perhaps with hydrogen peroxide (H2O2) and hypoxia inducible factors (HIF), cells increase transcription of the erythropoietin gene and subsequent production of the processed protein hormone. Basal physiologic levels range from approximately 6 to 32 U/L.(Van Dyke 1961) Serum levels of the hormone may transiently increase by a thousand-fold.
Erythropoietin has multiple actions. (Bahlmann 2004, Chong 2002, Rossert 2005) Its classic actions are well understood. Erythropoietin regulates erythrocyte production by stimulating progenitor cell proliferation and differentiation in the bone marrow. (Chong 2002, Ingley 2004) It also decreases erythrocyte apoptosis (cell death).(Chong 2002, Polenakovic 1996, Ratajczak 2001, Schwartz 1992) Less well understood are the roles erythropoietin may play either directly or indirectly in angiogenesis (blood vessel formation), e.g., wounds and the female productive tract (Chong 2002, Haroon 2003, Yasuda 1998, Zwezdaryk 2007) and the increase in thrombogenic properties of vascular endothelium.(Fruste 2002) Even less well understood are the proliferative effects it has on other tissues such as the bone marrow (stroma parenchyma) and tumors.(Lai 2005, Shiozawa 2010)
Erythropoietin activity is mediated through the classic erythropoietin receptor and perhaps non-classic receptor(s).(Rossert 2005, Sawada 1987, Szenajch 2010) Binding of the erythropoietin receptor by erythropoietin results in phosphorylation of Jak2 (janus kinase 2), which in turn activates other intracellular pathways STAT (signal transducer and activator of transcription), PI3K–Akt (phosphatidylinositol-3/Akt), and Ras/MAP (mitogen-activated protein) kinase.(Arcasoy 2005, Chong 2002, Kirschner 2000, Liang 2010, Pfeffer 2008, Ratajczak 2001, Solar 2008b, Yamazaki 2004) The expression of erythropoietin receptors on erythroid progenitor cells is well known. (D’Andrea 1989, Jones 1990, Winkelman 1990) Less well appreciated is the presence of erythropoietin-binding receptors on other tissues including cardiac myocytes, macrophages, neurons, vascular endothelial cells (Anagnostou 1994, Digicaylioglu 1995, Haroon 2003, Masuda 1993, Wright 2004), and cancers/cancer cell lines (bone sarcoma, breast, cervical, colon, gastric, head-neck [squamous cell], hepatoblastoma, melanoma, ovarian, pediatric, renal, retinal, and uterine (Acs 2001, 2002, 2003, Arcasoy 2003, 2005, Batra 2003, Liang 2010, Ribatti 2003, Selzer 2000, Solar 2008a, Szenajch 2010, Westenfelder 2000, Yasuda 2001).
Several forms of recombinant human erythropoietin have been developed (Table 1). (Jelkmann 2010, NKF Position Paper 1989, OTA 1990, Schellekens 2009) They differ in their carbohydrate structure. The most common species are erythropoietin-alpha and beta. The pharmacokinetic half-life of these products is six to eight hours after IV injection (Halstenson 1991). Because the pharmacodynamic response on the bone marrow is prolonged, dosing regimens vary from three times weekly to once weekly. Dosing via the intravenous route may require ~ 10-25% more drug for the same hematologic effect compared to subcutaneous administration. (Besarab 1992, Kaufman 1998, Paganini 1995) The erythropoietin molecule has been modified by the addition of 2 N-linked carbohydrate chains to form darbepoietin. The additional sialic acid residues decrease pharmacokinetic clearance by the body and permit weekly and semi-weekly dosing. (Egrie 2001, MacDougall 1999)
More recently, the erythropoietin molecule has been modified by the addition of a methoxy-poly-ethylene glycol polymer chain (pegylation) via a succinimidyl butanoic acid linker (MacDougall 2005). These changes further decrease pharmacokinetic clearance by the body and permit weekly and even monthly dosing. (MacDougall 2005) Although the molecular modifications decrease the affinity of the compound for the erythropoietin receptor in vitro, the increased residence time results in increased exposure of the compound to the erythropoietin receptor and increased erythropoietin-type activity in vivo. (Agoram 2008, El-Komy 2011, MacDougall 2003-abstract, 2005)
Molecules that activate the erythropoietin receptor or downstream pathways are under development. (Bugelski 2008 A and B, Johnson 1998, MacDougall 2008, Perez-Ruixo 2009, Sathyanarayana 2009, Sytkowski 1998, 1999, Wrighton 1996, 1997; Patents including #5,767,078, #5,773,569, #5,830,851, and #5,986,047 and patent applications including #20100249032.) These may be fusion proteins, erythropoietin dimers, truncated erythropoietin molecules, mimetic antibodies, or other small molecular entities. Others, such as GATA, may activate the receptor itself along with other hemoglobin synthesis genes. (Chiba 1991, Gregory 1999) Still others may activate/inactivate related pathways involving hypoxia-inducible transcription factor or hematopoietic cell phosphatase. (Bernhardt 2007, Del Vecchio 2010, Liu 2007) Phase III studies (Emerald 1 and 2, Pearl 1 and 2) have been conducted with peginesatide (formerly hematide), a pegylated peptidic erythropoiesis stimulating agent. (Affymax Analyst Day Handout 12/12/2010, Macdougall 2008, 2009, Stead 2006, Woodburn 2010)
Table 1: Erythrocyte Stimulating Agents
Recommended starting doses of erythropoietin (50 U/kg) results in serum erythropoietin levels that are supraphysiologic for many hours to days (Figure 1). (Brockmoller 1992) The supraphysiologic exposure (area-under-the-curve above) is greater in patients dosed via the intravenous route than via the subcutaneous route (Figure 1). (Brockmoller 1992) The supraphysiologic exposure is greater with higher dosing (Figure 2). (McMahon 1989) There are similar findings with the starting dose of darbepoetin (0.45 mcg/kg) and pegylated erythropoietin (0.6 mcg/kg) although the residence time is longer and the peak serum levels occur later with subcutaneous dosing. (Allon 2002, FDA darbepoietin review-pharmacokinetic section, FDA pegylated erythropoietin review-pharmacokinetic section, Locatelli 2007)
Figure 1: Serum levels of erythropoietin after a single dose 50 U/kg by route of administration (Brockmoller 1992)
Panel A Intravenous Dose Panel B Subcutaneous Dose
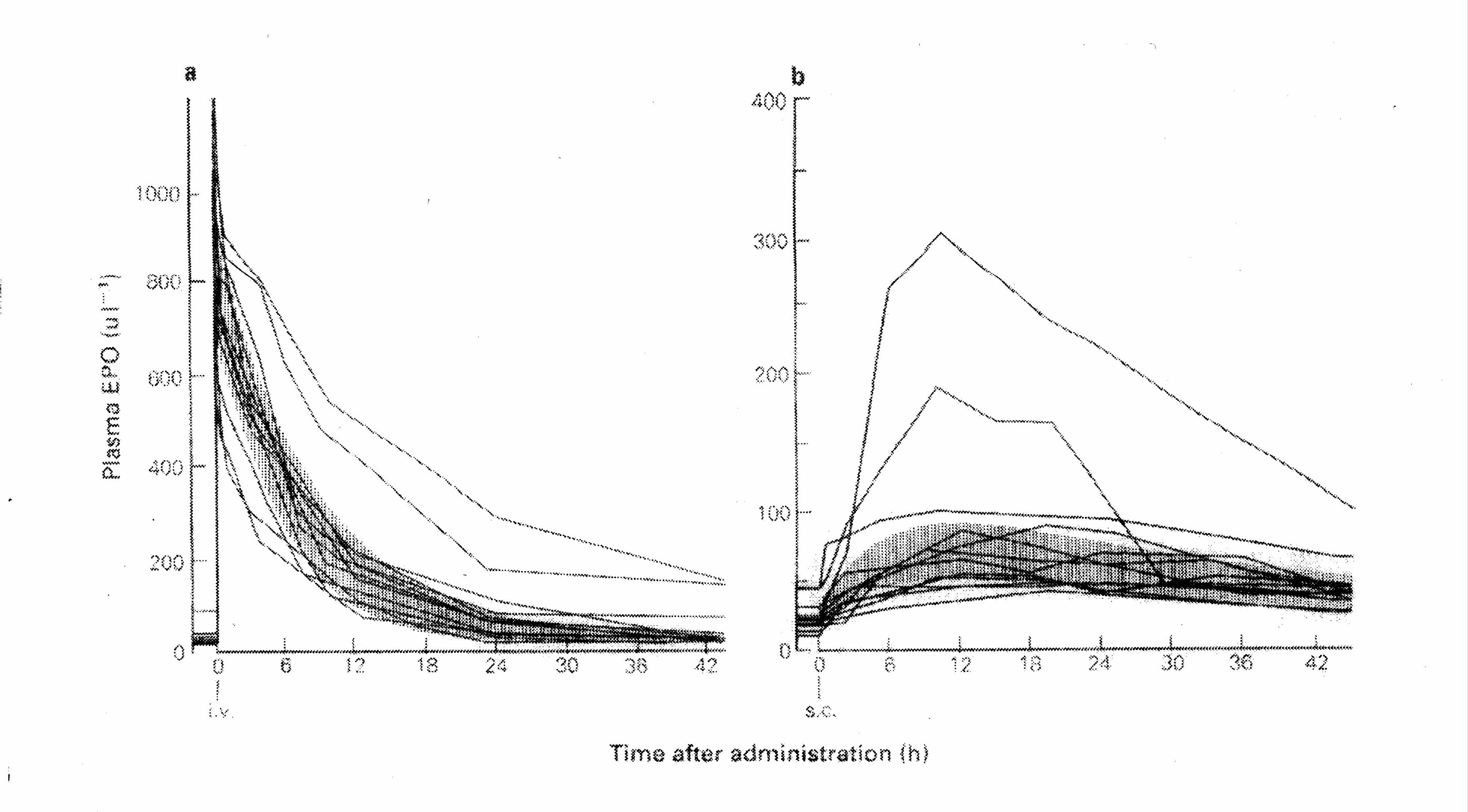
Figure 2: Serum levels of erythropoietin after a 300 U/kg intravenous dose on days 1 and 10 (McMahon 1989)
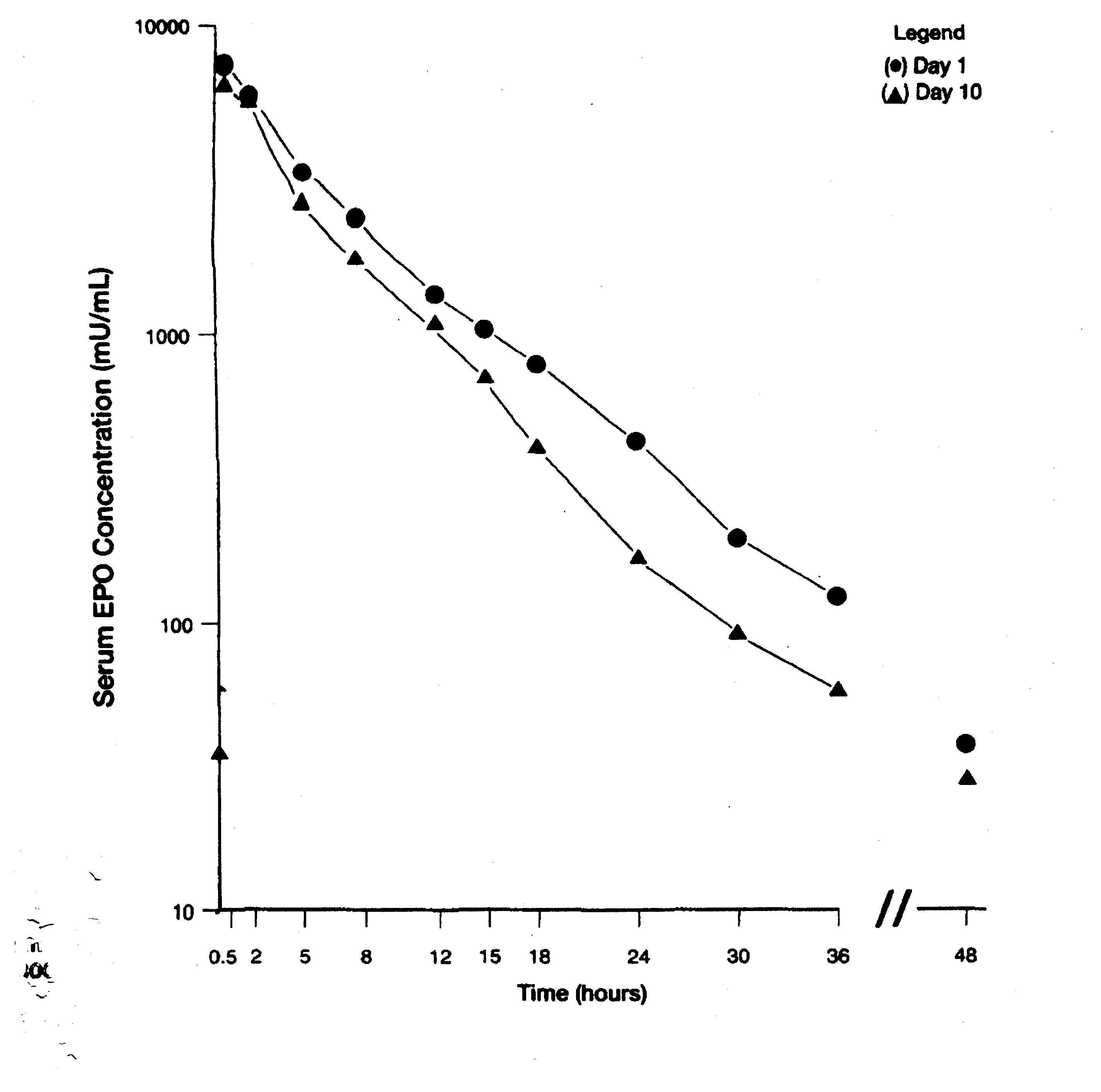
Basal physiologic levels of erythropoietin range from approximately 6 to 32 U/L.
B. Disease Summary
The kidneys are responsible for multiple aspects of physiologic homeostasis. They do this by maintaining acid-base balance, maintaining electrolyte balance, regulating whole body water content, filtering water soluble toxins, retaining/preventing the loss of re-usable biochemical entities, e.g., glucose and proteins including albumin, activating vitamin D to facilitate calcium absorption, and mitigating hypoxia. Renal disease may impair these functions.
Kidney damage may manifest itself with urinary protein loss, abnormal urinary sediment with casts and/or blood cell elements (erythrocyte or leukocytes), or structural changes present on medical imaging (scarring, size reduction, and/or cystic changes) even before decreased glomerular filtration is detected. (Levey 2009) In Stage 1 disease, the glomerular filtration rate (GFR) is normal or increased (≥ 90 mL/min/1.73 m2), but there are other chronic pathologic findings of damage. In Stage 2 disease, the glomerular filtration rate is minimally decreased (60-89 mL/min/1.73 m2) and there are other chronic pathologic findings of damage. In Stages 3 and 4, the glomerular filtration rates are minimally decreased to 30-59 mL/min/1.73 m2 and 15-29 mL/min/1.73 m2. In Stage 5 disease, the glomerular filtration rate is less than 15 mL/min/1.73 m2 and/or dialysis is required for management of electrolytes, fluids, and/or uremic toxins. (For claims purposes, A further distinction is made in patients with endstage renal disease via the ICD-9 codes: Stage 5 585.5 for those with a GFR less than 15 ml/min1.73 m2 and not on dialysis and Stage 6 585.6 for those on chronic dialysis.)
Symptoms, primarily attributable to uremia, reduced fluid clearance, urinary protein loss, and secondary hypertension may present when glomerular filtration is below 30 mL/min/1.73 m2 and become more noticeable with further declines in renal function. Symptoms include alterations in sleep patterns, anorexia, bruising, chest discomfort, dysgeusia (abnormal taste), dyspnea, edema, fatigue, gastrointestinal bleeding, itching, impaired cognitive function, insomnia, muscle cramps, nausea, and changes in micturition patterns. With the progression of renal disease, patients may lose physical function and independence. Cross-sectional Medicare claims data reveal that use of assistive devices for walking (canes, walkers, wheelchairs) is 16.9% in the pre-dialysis chronic kidney disease population and 32.5% in the incident dialysis population. (USRDS 2008, 2009) The data also reveal that a walking disability (abnormal gait, difficulty walking, fall history) is present in 19.2% of incident dialysis patients and that 40.1% of incident dialysis patients go on to develop a new walking disability during the first year on dialysis. (USRDS 2008, 2009)
Chronic kidney disease (pre-dialysis and end-stage renal disease requiring dialysis) has become more common in the U.S over time. Cross-sectional laboratory data (persistent microalbuminuria [> 30 mg/g creatinine] and calculated glomerular filtration derived from serum creatinine values and the Modification of Diet in Renal Disease equation) from National Health and Nutrition Examination Surveys (NHANES) II (1988-1944) and III (1999-2004) revealed an increase in the prevalence of pre-dialysis kidney disease in the general adult (≥ 20 years) population. (Coresh 2007) The largest prevalence increases were found in patients with Stage 2 disease (2.7% to 3.2%) and Stage 3 disease (5.4% to 7.7%). Cross-sectional claims data revealed an increase in pre-dialysis kidney disease from 2.9% to 7.9% whereas data from the Medical Evidence form (2728) revealed an increase in end-stage renal disease (ESRD) requiring dialysis and/or transplantation from 0.8% to 1.1% in the general Medicare population from 1996 to 2006. (USRDS 2008)
The demographics of the end-stage renal disease population in the U.S. have changed over time. The adjusted incident rate for patients 19 years and under has remained relatively low and stable at 13-15/million from 1988 to 2006 (USRDS 2008, 2009). The adjusted incident rate for patients 20 to 44 years of age has increased minimally and gradually from 97/million to 127/million. By contrast the adjusted incident rate for older adults has increased significantly: a) almost double (363/million to 625/million) for patients 45 to 64 years of age, b) more than double (668/million to 1452/million) for patients 65 to 74 years of age, and c) tripled 517/million to 1744/million for patients 75 years and older.(USRDS 2008, 2009) By contrast, ESRD prevalence is highest for patients aged 45 to 64 years of age and the adjusted prevalence rate is highest for patients aged 65 to 74 years of age and reflects the overall mortality associated with age and increased mortality especially within the first year of dialysis respectively.(USRDS 2008, 2009)
The causes of end-stage renal disease in the U.S. have also changed over time. Although the major causes of ESRD (diabetes-related, hypertension, glomerulonephritis, and cystic kidney disease) have remained the same, their relative importance has changed. The incidence of diabetes-related and hypertension-related renal disease has increased markedly. Much of the increase in diabetes-related renal disease may reflect the underlying macrovascular disease and hypertension associated with the metabolic derangement of Type 2 diabetes (and not the classic microvascular renal disease associated with Type 1 diabetes). By contrast, glomerulonephritis was the most common cause of renal disease in the prevalent population in the early 1980s, and currently both glomerulonephritis and cystic kidney disease are disproportionately represented in the prevalent population when compared to the incident population. This reflects the increased mortality associated with diabetes-related renal disease and hypertension as well as the age-of-onset associated with these disorders. (Churchill 1992)
The current end-stage renal disease population is currently older and has more co-morbid disease (especially antecedent hypertension, type 2 diabetes, and atherosclerosis-lipids dysfunction). (Knauf 2009, NKF Position Paper 1989, Pedersen 2009, Sorace 2011, USRDS 2008, 2009) Annual mortality rates are higher for older patients. (USRDS 2010) Mortality rates during the first year on dialysis have remained unchanged. (USRDS 2008, 2009) Survival in that first year is approximately 60% in the overall incident dialysis population and 40% in patients who are unable to walk. The five-year survival in the dialysis population is approximately 30%. (USRDS 2008, 2009) Cardiovascular-related mortality, which has fluctuated between 79 deaths/103 patient-years in 1991, 94.1 deaths/103 patient-years in1999, and 72.1 deaths/103 patient-years in 2006, is responsible for approximately 50% of overall mortality. (USRDS 2008, 2009, 2010)
Although the number of renal transplants has increased over time, both age and cause of renal disease are factors in whether a patient (with onset of ESRD less than 70 years of age) has received a renal transplant within three years of ESRD registration and these demographic features have changed little since 1991. (USRDS 2008, 2009, 2010) Patients with cystic kidney disease (~ 45-50%) and glomerulonephritis (~35-40%) are more likely to receive a transplant than those with hypertension and diabetes-related renal disease (~ 12-18%). Younger patients (aged < 20 years; ~70%) are more likely to receive a transplant than older patients (age 20-39 years; 47% declining to 31%; age 40-59 years; 25% declining to 18%, and age 60-69 years; 6% increasing to 9%).
Anemia in Renal Disease-Etiology
There are multiple causes of anemia in patients with renal disease. There is decreased red cell production and increased red cell loss. Uremia reduces erythrocyte survival and suppresses hematopoietic cell production in the bone marrow. (Delwiche 1986, Fukushima 1986, Radtke 1980) Uremia may cause hemorrhagic bleeding, often from the gastrointestinal tract. (Andrassy 1985, Kang 1990, 1993, 1999, Rabiner 1972, Schiller 1989) The hemodialysis procedure and the filters used result in frank blood loss and decreased red blood cell survival. (Handelman 2010) Because of anorexia and dietary restrictions, oral intake of important nutrients, e.g., iron (Fe), may be inadequate. (DeVita 2003, Donnelly 1990, Kotaki 1997, van Wyck 1989) Aluminum (Al), which may be used for phosphate binding and as an antacid to reduce occult bleeding, may have a direct toxic effect on hematopoiesis and an indirect effect impairing iron metabolism. (Bia 1989, Caramelo 1995, Donnelly 1990) Erythropoietin deficiency in many patients with renal disease reduces marrow stimulation of hematopoietic cells although endogenous production (made by the body) of erythropoietin is relatively preserved in some types of renal disease, e.g., polycystic kidney disease. Erythropoietin production and utilization by the body may also be decreased in the setting of other nutritional co-factors, e.g., iron and vitamins. (Altallah 2006 Amato 2005, DeVita 2003, Goicoechea 1998, Keven 2003, MacDougall 1995)
There may be resistance to erythropoietin, whether endogenous (made by the body) or exogenous (made outside the body) in the setting of dialysis inadequacy, dysplastic marrow, occult or frank inflammation, infection, anti-erythropoietin antibodies, putative receptor defects, and putative anti-erythropoietin receptor antibodies. (Agarwal 2008, Boven 2005, Casadevall 1996, de la Chapelle 1993, Di Iorio 2003, Drueke 1990, Elliot 2009, Howman 2007, Ifudu 1996, Jacob 2005, Kalantar-Zadeh 2003, Kaysen 2003, 2006, Kilpatrick 2008, Khankin 2010, Kralovics 1997, MacDougall 1995, Markson 1956, Movilli 2003, Nassar 2002, Pedersen 2009, Radtke 1981, Rossert 2007, Ryan 2006, Schellekens 2006, Schreiber 1996, Solomon 2010, Szczech 2008, Wallner 1981, Zappacosta 1982)
Hyperparathyroidism, usually present as a secondary phenomenon to hypocalcemia in renal disease, has been postulated to cause anemia via several mechanisms including specific type of marrow fibrosis (osteitis fibrosa cyctica) impairing hematopoietic cell production. (Bhadada 2009, Gallieni 2000, Grutzmacher 1983, Massry 1983, McGonigle 1984, Rao 1993) Medications used in the management of renal disease, e.g., erythropoietic stimulating agents may cause (semi-)reversible marrow fibrosis with different pathologic features. (Akada 2010, Bader 1992, Barosi 2005, Dokal 1989, Epogen label, Gallieni 2000, Hussein 2007, Kakumitsu 2005, Kennedy 2006, Lacout 2006, Levine 2005, Reilly 1997, Shiozawa 2010, Snide 2008, Tulliez 1989, Wernig 2006)
In addition, many patients with renal dysfunction have co-morbid conditions that are the underlying cause(s) of their anemia. For example, cytokines associated with the anemia of chronic disease may impair hematopoietic nutrient utilization, erythropoietin production, and erythropoietin efficacy. (Means 1992, Pedersen 2009) The presence of a mild anemia in type 2 diabetes is only now being recognized and may be a variant of the anemia of chronic disease. (Ishimuura 1998, Thomas 2003)
Anemia can be attributed to renal dysfunction only when there is significant renal dysfunction (Figure 3). (Radtke 1979) Mild anemia (mean hematocrit ~ 37 volume %) may be present when the glomerular filtration rate is between 30 and 40 ml/min/173 m2. It is more common (mean hematocrit ~ 33 volume %) when the clearance is between 20 and 30 ml/min/173 m2. Modest anemia (mean hematocrit ~ 30 volume %) is present when the clearance is between 10 and 20 ml/min/173 m2.
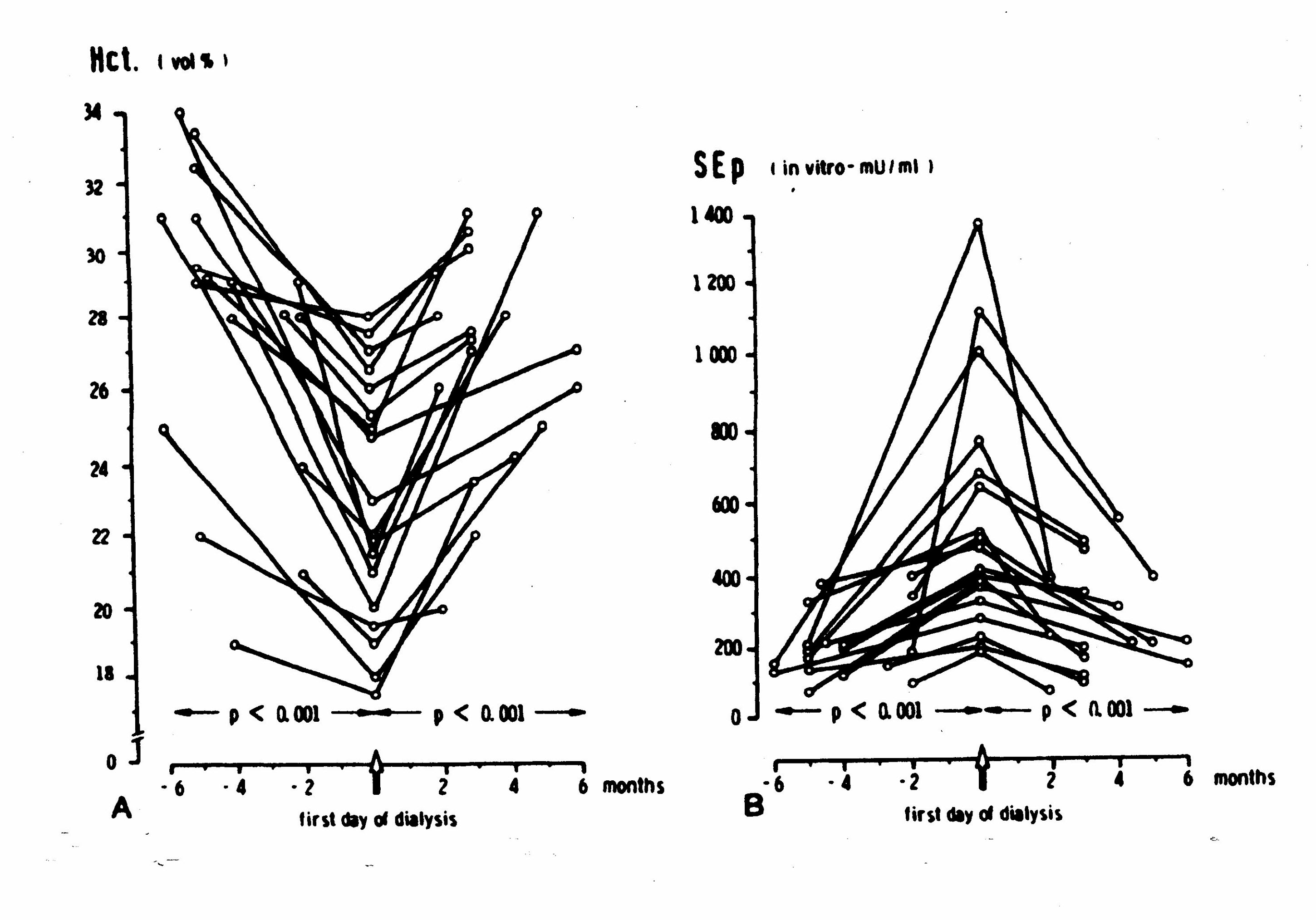
Longitudinal data demonstrate that hematocrit levels decline in the 6 months prior to the initiation of dialysis and rebound, without exogenous erythropoietin, in the months immediately subsequent to the initiation of dialysis (Figure 4; Panel A). (Erbes 1978, Radtke 1979, 1980) Concomitant longitudinal data show that endogenous erythropoietin levels rise in the 6 month prior to the initiation of dialysis and decline in the months immediately subsequent to the initiation of dialysis (Figure 4; Panel B). (Radtke 1979) In the six to twelve months after the initiation of dialysis, both hematocrit and endogenous erythropoietin levels decline and remain low in most patients-even when dialysis is adequate. (Radtke 1979) Select patients, including those with polycystic kidney disease, retain some erythropoietin-production capacity. (Brown 1980, Eckardt 1991, Koch 1979, Radtke 1977, Ross 1994, Zeier 1996) Such data suggest that the uremia is the primary underlying etiologic agent for anemia in the pre-dialysis patient and that the kidney (and extra-renal tissue) respond to the challenge of anemia with increased production of the erythropoietin hormone in the pre-dialysis patient. Consistent with classic hormone feedback loops, the removal/reduction of the anemia-causing toxins, via dialysis and other renal management measures, decreases the need for erythropoietin secretion. Then, with continued deterioration of the renal parenchyma over time, the functional capacity for both filtration and erythropoietin production is lost (for most patients). The hormonal feed-back loop ceases to function in patients with well-established chronic renal failure. At this stage, erythropoietin deficiency becomes a major underlying cause of anemia.
Figure 3: Hematocrit Level and Renal Function (Radtke 1979)
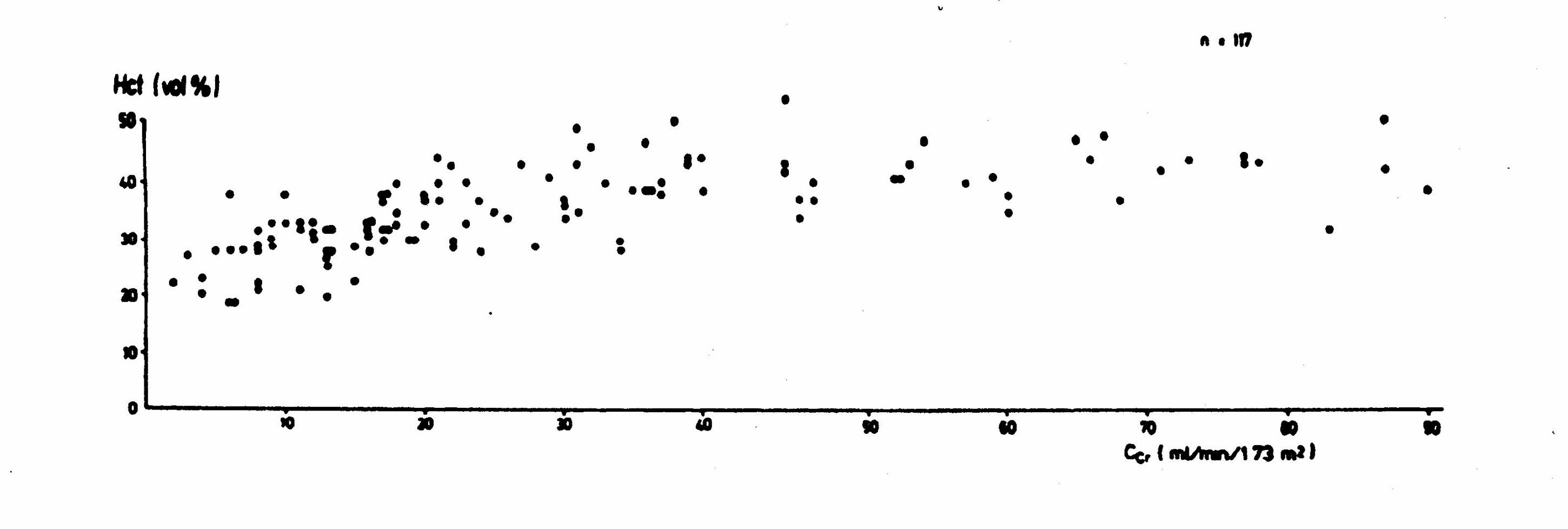
Figure 4:Hematocrit, Erythropoietin, and Renal Function (Radtke 1979)
Panel A Changes in Hematocrit in Response to Uremic State
Panel B Changes in Erythropoietin in Response to Hematocrit and Uremic State
Anemia in Renal Disease-Demographics Features
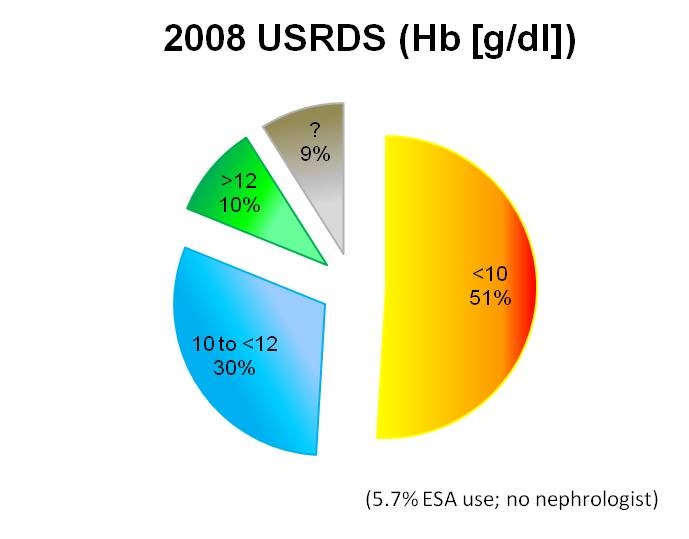
The severity of anemia in end-stage renal disease patients appears to have changed over time. Secular changes suggest that hemoglobin/hematocrit levels are currently higher in ESA-naïve patients. Data from the 2008 USRDS annual publication suggest that 51% of incident ESRD patients have hemoglobin levels < 10 g/dl (hematocrit ~ 30 volume%) (and 9% unknown) whereas 1990 Congressional-Office of Technology (OTA) data indicate that 74% had hematocrit levels < 30 volume % (hemoglobin ~10 g/dl) (Figures 5 and 6). Forty-one percent of these had hematocrit levels 25 to < 30 volume %; thirty percent had hematocrit levels from 20 to < 25 volume %; three percent had hematocrit levels under 20 volume %. These differences may reflect changes in patient management, patient composition, and/or some other unknown factor. (Eggers 2000)
Figure 5: Level of Anemia Prior to Significant ESA Use in U.S.

Figure 6: Level of Anemia in Current Pre- dialysis Patients (Population not treated by a nephrologist. ESA use in 5.7%)
Historical Treatment of Anemia
It was long presumed that anemia contributed to the fatigue and poor level of functioning in renal disease and that therapeutic intervention was warranted although the level at which anemia requires intervention is not well established. By tradition, patients have been transfused with packed red blood cells (PRBCs) at the hemoglobin level of 7 or 8 g/dl to avoid symptoms and physiologic complications. (Churchill 1992, Ibrahim 2008, 2009) A transfusion of two or more units of PRBCs would result in an increase of at least 2 g/dl of hemoglobin (6 volume % units of hematocrit). Most of these practices, however, are based on empiric observations and not clinical trials. Anemia in renal disease prior to the development of ESAs was primarily treated with transfusions. In 1992, in the year post initiation of dialysis, approximately 19% of patients received a single transfusion, 8% received two transfusions, and 7% received three or more transfusions. (USRDS 2008). Other therapeutic interventions included androgens, e.g., nandrolone and nutrients, e.g., iron (oral or intravenous).
In 1906, erythropoietin was identified as a regulatory hormone for red cell production and, in 1957, its source identified as the kidneys. (Gurney 1957, Reissman 1960) Commercialization was limited by the availability of processes for extraction, replication, and purification of the protein. In the 1980s, with the advent of recombinant technology, several companies, e.g., Amgen and Genetics Institute, attempted commercialization of a therapeutic product. Amgen and the Genetics Institute received Orphan Drug status from the FDA for their respective products, erythropoietin α and erythropoietin β. (Asbury 1991) Amgen partnered with Ortho Pharmaceutical Company. Amgen retained marketing rights for erythropoietin in the U.S. dialysis population. (Coster 1992, NKF Position Paper 1989) Genetics Institute partnered with Chugai (Japan) and Chugai-Upjohn with the latter holding the marketing rights to erythropoietin in the U.S.(Coster 1992, NKF Position Paper 1989) In 1989, the FDA approved recombinant erythropoietin α to manage anemia decrease transfusions in dialysis patients and in pre-dialysis patients in whom hemoglobin levels were less than < 10 g/dl. In 2001, darbepoetin alpha (α) was approved by the FDA to increase hemoglobin.
Over time, ESAs became used in a greater proportion of dialysis patients, a greater proportion of pre-dialysis patients, and in renal patients with less severe anemia (Figure 7). (Besarab 1993, USRDS 2008) The dose of ESAs has increased over time (Figure 8). (Collins 1997, USRDS 2008, 2009) Dosing in the U.S. differs from that of Europe, where dosing is approximately 50% less for equivalent hemoglobin levels (Tables 29, 30, and 31). (Burton 2000, Jacob 2005, Pisoni 2004, Richardson 2009)
Figure 7: Change in Hemoglobin Levels and ESA Use over Time
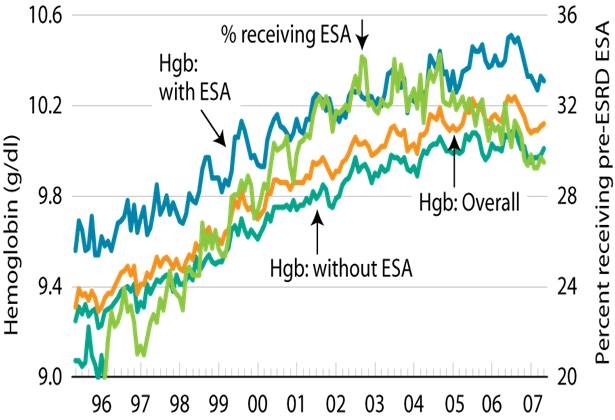
Figure 8: Change in ESA Doses over Time
III. History of Medicare Coverage
The end stage renal disease program in Medicare was established by the Social Security Amendments of 1972, Public Law 92-603, Section 299I (1972). Medicare coverage of dialysis typically started during the fourth month of dialysis. Services and items covered by the program include dialysis procedures whether in-patient or out-patient, dialysis supplies, blood transfusions, transplantation, some transplantation-related costs, and drugs associated with dialysis, e.g., heparin and ESAs. These medications are paid under Medicare Part B.
There is no national coverage determination (NCD) concerning the use of ESAs in beneficiaries with renal disease treated with dialysis and beneficiaries with renal disease in pre-dialysis stages.
A. Current Request
On June 16, 2010 CMS accepted a formal request for a NCD with respect to Medicare coverage of ESAs for treatment of chronic kidney disease (CKD) and dialysis-related anemia from Mr. Dennis Cotter, President, Medical Technology & Practice Patterns Institute (MTPPI.) His letter is available at the following link: http://www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/id245.pdf.
B. Benefit Category
Medicare is a defined benefit program. An item or service must fall within a benefit category as a prerequisite to Medicare coverage §1812 (Scope of Part A); §1832 (Scope of Part B) and §1861(s) (Definition of Medical and Other Health Services) of the Act. ESAs fall within the benefits categories specified in §1861(s)(2)(O)of the Social Security Act.
IV. Timeline of Recent Activities
September 2009
CMS commissioned a technology assessment (TA) to search the literature for ESA clinical trials.
November 2009
CMS commissioned a TA that would describe ESA utilization in Medicare beneficiaries with renal disease. The information was presented at the March 24, 2010 MEDCAC.
June 16, 2010
CMS accepted a formal request for an NCA to evaluate erythropoiesis stimulating agents (ESAs) for treatment of anemia in adults with CKD including both patients on dialysis and patients not on dialysis. A tracking sheet was posted on the web site and the initial 30 day public comment period commenced. CMS commissioned a technology assessment to delineate the role and impact of blood transfusion on renal transplantation.
July 16, 2010
The initial 30 day public comment period ended. Nine timely comments were received.
January 19, 2011
CMS held a Medicare Evidence Development and Advisory Committee (MEDCAC) meeting to discuss the role and impact of blood transfusion on renal transplantation. (www.cms.gov/ medicare-coverage-database/details/medcac-meeting-details.aspx?MEDCACId=57&bc =BAAQAAAAAAAA&; accessed January 21, 2011.)
March 16, 2011
Proposed decision memorandum posted on the CMS website.
V. FDA Status
A. In 1989, the FDA approved erythropoietin-alpha for the treatment of anemia in renal disease. It was the first erythropoiesis stimulating agent (ESA) approved by the FDA.
B. In 1993, the FDA approved erythropoietin-alpha for the management of the anemia due to myelosuppressive cancer chemotherapy of solid tumors.
C. On September 17, 2001, the FDA approved the long-acting erythropoietin analogue, darbepoetin, to increase hemoglobin in renal disease patients. (www.accessdata.fda.gov/drugsatfda_docs/appletter/2001/darbamg091701L.htm, www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm080442.htm, www.accessdata.fda.gov/drugsatfda_docs/label/2001/darbamg091701LB.htm; accessed July 19, 2010.)
D. On July 19, 2002, the FDA approved darbepoetin for the management of the anemia due to concomitantly administered chemotherapy for non-myeloid cancer. See www.accessdata.fda. gov/drugsatfda_docs/appletter/2002/darbamg071902L.htm and www.accessdata.fda.gov/drugsatfda_docs/label/2002/darbamg071902LB.pdf. (Accessed July 19, 2010.)
E. In 1997, 2004, 2005, 2007, and 2008, ESA product labeling underwent substantial revisions. (Accessed July 19, 2010.)
1-Epogen/Procrit
www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm080580.htm (revised pediatric use section for renal disease; 4 studies in dialysis patients (EPO 9118 single arm n = 74, EPO 8702 single arm n = 5, EPO 8905 double-blind n = 10, EPO 9902 double-blind n = 112 )
www.accessdata.fda.gov/drugsatfda_docs/label/2000/epoamg072699lb.pdf (request for literature on pharmacokinetic data in neonatal use)
www.accessdata.fda.gov/drugsatfda_docs/appletter/biologics/2004/103234-5033ltr.pdf (phase 4 commitment N93-004 to assess ESA effect on solid tumor growth completed; agreement made with 1993 approval; agreement to conduct survival/time to tumor progression study in metastatic breast cancer patients; update warnings and precautions sections for cancer patients; dear doctor letter.)
www.accessdata.fda.gov/drugsatfda_docs/nda/2004/103234s5033.pdf (review of BEST trial; advised recent proposed label changes not acceptable; request for information on thrombosis-vascular events, tumor progression, and cancer treatment response rates in randomized, placebo controlled studies with patients with a single tumor type and anti-cancer treatment regimen.
www.accessdata.fda.gov/drugsatfda_docs/appletter/biologics/2004/103234_5053ltr.pdf (acknowledgement that study PR99- 11-034/044, a study of anemia and quality-of-life children with solid tumors, Hodgkin s disease, ALL, or NHL and undergoing myelosuppressive chemotherapy, has been completed, but not yet received for review; request for deferred studies in pediatric cancer patients five years and under)
www.accessdata.fda.gov/drugsatfda_docs/nda/2004/103234s5053.pdf (review of several studies in cancer patients for weekly dosing and hemoglobin, time to transfusion, and quality-of-life parameters; survival curve in PR98-27-008 appears to diverge after approximately 500 days and favors the placebo arm)
www.accessdata.fda.gov/drugsatfda_docs/label/2004/103234_5053_Epogen_lbl.pdf (alternative weekly dosing was added for cancer patients)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2004/103234_5076ltr.pdf (acknowledgement of submission of literature search for pharmacokinetic information on use in neonates in response to a 1997 commitment)(use in children based on literature, renal: Campos 1992, Montini 1990, Offner 1990, Muller-Wiefel 1988, Sharer 1993; HIV: Mueller 1994, Zuccotti 1996; cancer: Beck 1995, Bennettts 1995.)
www.accessdata.fda.gov/drugsatfda_docs/nda/2004/103234s5076_AP_PKG.pdf (literature submitted: Kling and Widness 1992 case report of infant with urinary tract obstruction, Widness 1996 seven premature infants and ten adults)
www.accessdata.fda.gov/drugsatfda_docs/label/2004/103234_5076lbl.pdf (two studies above included in label)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/103234s5093ltr.pdf (added information about pure red cell aplasia for renal disease section, update renal section of patient insert, distribute dear doctor letter to hematology-oncology care providers)
www.accessdata.fda.gov/drugsatfda_docs/nda/2005/103234s5093_AP_PKG.pdf (pure red cell aplasia case reports in system and packaging issues resulting in administration errors viewed)
www.accessdata.fda.gov/drugsatfda_docs/label/2005/103234s5093lbl.pdf (IV route recommended for hemodialysis patients to possibly reduce risk of pure red cell aplasia)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2006/103234s5104_LTR.pdf (unspecified label and patient insert changes)
www.accessdata.fda.gov/drugsatfda_docs/label/2006/103234s5104_LBL.pdf (unspecified label changes)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2007/103234s5122ltr.pdf (increased warnings and precautions; removed quality of life claims, requested substantiation of any patient-related outcome (PRO) claims in accordance with the FDA guidance and to be received by June 15, 2007)
www.accessdata.fda.gov/drugsatfda_docs/label/2007/103234s5122lbl.pdf (addition of boxed warning for increased risk of death, cardiovascular events, thrombo-embolic events, tumor progression; include information delineating increased risk with use in renal and HIV patients; remove quality of life claims; clarify dosing strategies)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2007/103234s5158ltr.pdf (strengthen box label warnings and send dear doctor letter)
www.accessdata.fda.gov/drugsatfda_docs/label/2007/103234s5158lbl.pdf (cardiovacular-thombotic risk for renal and surgical patients more clearly outlined in boxed warning)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2008/103234s5163ltr.pdf (typographical error in table 1 in warning section to be corrected)
www.accessdata.fda.gov/drugsatfda_docs/label/2007/103234s5163lbl.pdf (addition of boxed warning for increased risk of death, cardiovascular events, thrombo-embolic events, tumor progression; include information delineating increased risk with use in renal and HIV patients; remove quality of life claims; clarify dosing strategies)
www.accessdata.fda.gov/drugsatfda_docs/label/2008/103234s5164lbl.pdf (unspecified changes in label and patient insert)
2-Darbepoetin
www.accessdata.fda.gov/drugsatfda_docs/appletter/2004/103951_5069ltr.pdf (thrombosis and tumor progression; dear doctor letter)
www.accessdata.fda.gov/drugsatfda_docs/label/2004/103951_5069lbl.pdf (thrombosis and tumor progression; label change)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/103951s5096ltr.pdf (pure red cell aplasia; dear doctor letter)
www.accessdata.fda.gov/drugsatfda_docs/label/2005/103951s5096lbl.pdf (pure red cell aplasia; label change)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2006/103951s5088ltr.pdf (agreement to provide information on 20010145 in small cell lung cancer patients, DE 2001-0033 (PREPARE-CIA in chemotherapy patients, DE-2002-0015 (ARA-03) in breast cancer patients, SE-2002-9001 (DAHANCA-10) in head-and-neck cancer patients, FR-2003-3005 (GELA LNH-036B) large B-cell lymphoma patients, adverse events [12/2011])
www.accessdata.fda.gov/drugsatfda_docs/label/2006/103951s5088lbl.pdf (dosing regimen q3 weeks)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2007/103951s5139ltr.pdf (boxed label warning section for cardiovascular, thrombotic, and tumor growth potential; provide information on survival in cancer patients)
www.accessdata.fda.gov/drugsatfda_docs/label/2007/103951s5139lbl.pdf (increase severity of adverse event warnings in label)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2007/103951s5135ltr%20.pdf (allergic reactions with rubber stoppers for vials)
www.accessdata.fda.gov/drugsatfda_docs/label/2007/103951s5135LBL.pdf (allergic reaction; label change)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2007/103951s5164ltr.pdf (dear doctor letter with new label changes)
www.accessdata.fda.gov/drugsatfda_docs/label/2007/103951s5164lbl.pdf (change in label, package insert, patient information)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2007/103951s5169ltr.pdf (correction of typographical error in warning section)
www.accessdata.fda.gov/drugsatfda_docs/label/2007/103951s5169lbl.pdf (typographical error; label change)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2008/103951s5170ltr.pdf (includes data from DE 2001-0033 (PREPARE) and GOG191; dear doctor letter)
www.accessdata.fda.gov/drugsatfda_docs/label/2008/103951s5170lbl.pdf (label change to warnings and boxed warnings sections)
www.accessdata.fda.gov/drugsatfda_docs/label/2008/103951s5195PI.pdf (updated label)
www.accessdata.fda.gov/drugsatfda_docs/appletter/2009/103951s5211ltr.pdf (pure red cell aplasia in setting of hepatitis C treated with ribavirin and HIV and ribavarin and interferon; dear doctor letter)
www.accessdata.fda.gov/drugsatfda_docs/label/2009/103951s5211Lbl.pdf (updated warnings section for red cell aplasia in label)
F. In 2004, the FDA reviewed results of the Breast Cancer Erythropoietin Trial (BEST) and Henke studies. On May 4, 2004, the FDA convened a meeting of the Oncologic Drugs Advisory Committee May 4, 2004 to discuss safety issue for ESAs. The briefing information and transcript for the meeting is available at www.fda.gov/ohrms/dockets/ac/cder04.html#Oncologic. Later that year, concerns regarding an increased rate of tumor progression and increased mortality were incorporated into the precautions section of product labeling. (Accessed July 19, 2010.)
G. In February, 2006, the FDA issued a draft guidance for patient report outcomes (PRO). See www.fda.gov/OHRMS/DOCKETS/98fr/06d-0044-gdl0001.pdf and www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ucm118795.pdf. (Accessed July 19, 2010.)
H. On November 16 or 17 (sic), 2006, the FDA issued the first of a series of alerts regarding ESA safety.
I. On January 26, 2007, the FDA issued a “Dear Doctor Letter” regarding the use of ESAs for anemia management in the absence of chemotherapy. See www.fda.gov/medwatch/safety/2007/safety07.htm#Aranesp. (Accessed July 19, 2010.)
J. On February 16, 2007, the FDA notified healthcare providers of increased mortality and no transfusion decrease in a study in darbepoetin using cancer patients not receiving chemotherapy. See www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm152120.htm. (Accessed July 19, 2010.)
K. On March 9, 2007, the FDA notified healthcare providers of increased adverse events including death in four studies of cancer patients. The trials were studying ESA use in an off-label patient population, in an off-label dosing regimen, or with an unapproved ESA. See www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm152120.htm. (Accessed July 19, 2010.)
L. In March 2007, the FDA sent Amgen a letter requesting that Amgen, in a post-marketing commitment, reassess the data used to make patient report outcomes (PRO) in ESA labels in concordance with the principles laid out in the FDA draft PRO guidance document. Amgen agreed to remove quality-of-life claims (e.g., happiness, life satisfaction, and well-being) from ESA labels. Claims that could be considered would be limited to health-related quality of life claims (physical, psychological, and social functioning that reflect the impact of a disease and its treatment). The sponsor was to provide the information by June 15, 2007.
The FDA noted that the instruments for PRO claims must have content validity (documentation that the test items are derived from patient input and are appropriate, clinically meaningful, well-defined, specific to the target population/indication, interpretable, and comprehensive), construct validity, reliability, and the ability to detect change. If instruments are altered or used in different patient populations, they require re-validation. PRO instruments will not provide meaningful information unless they are used in adequately designed studies with blinding and prospective statistical analysis plans. Plans to address missing data and drop-outs must be made.
M. On September 11, 2007, the FDA convened a joint meeting of the Cardio-Renal Drugs Advisory Committee (CRDAC) and Drug Safety and Risk Management Advisory Committee to discuss safety issue for ESAs. The briefing information and transcript for the meeting is available at www.fda.gov/ohrms/dockets/ac/cder07.htm#CardiovascularRenal. (Accessed July 19, 2010.)
The FDA determined that on the basis of the documents submitted to the FDA by July 2007 that the PRO claims made in the label for erythropoietin were not adequately substantiated. Documents submitted subsequent to July 2007 were to be reviewed after the CRDAC meeting date.
N. On November 8, 2007, the FDA notified healthcare professionals of ESA label changes including black box warnings. The warnings noted the tumor growth and shortened survival in study patients with advanced breast cancer, head and neck cancer, lymphoid cancer, and non-small cell cancer in which the ESA was dosed in an attempt to reach a hemoglobin of ≥ 12 g/dl. The warnings noted that ESAs, in the setting of cancer, should be used only when the anemia was due to the chemotherapy and should be discontinued with the cessation of chemotherapy. The notice provided information on management of poor responders to ESAs. See www.fda.gov/safety/medwatch/safetyinformation/safetyalertsforhumanmedicalproducts/ucm152274.htm. (Accessed July 19, 2010.)
O. On November 8, 2007, the FDA notified healthcare professionals of ESA label changes including black box warnings. The warnings noted that maintaining hemoglobin levels higher than 12g/dl increased the risk of death and other adverse events in patients with chronic renal failure. The notice provided information on management of poor responders to ESAs.
P. On November 12, 2007, the FDA approved methoxy polyethylene glycol-epoetin beta for the treatment of anemia in renal disease. See www.accessdata.fda.gov/drugsatfda_docs/nda/2007/125164TOC.cfm and www.accessdata.fda.gov/drugsatfda_docs/label/2007/125164lbl.pdf. (Accessed July 19, 2010.)
Q. On January 3, 2008, the FDA notified healthcare professionals of additional studies demonstrating tumor growth and shortened survival in patients with breast cancer (Preoperative Epirubicin Paclitaxel Aranesp Study [PREPARE]; Germany; n = 733) and cervical cancer (National Cancer Institute Gynecologic Oncology Group [COG-19] [sic GOG 191]; chemotherapy and radiation; 109 of 460 enrolled) after being notified by Amgen on November 30 and December 4, 2007 respectively. Enrollment was stopped early in the NCI study because of an imbalance in serious blood clots. Healthcare professionals were encouraged to review ESA risks with patients. See www.fda.gov/safety/medwatch/safetyinformation/safetyalertsforhumanmedicalproducts/ucm152274.htm. (Accessed July 19, 2010.)
(PREPARE information filed to clinicaltrials.gov/ct2/show/NCT00544232 without subsequent change on October 15, 2007. GOG-191 recruitment closure filed to clinicaltrials.gov/archive /NCT00017004/2007_08_06 on August 6, 2007.) (Accessed July 19, 2010)
R. On August 14 and 15, 2008, the FDA convened a meeting of the Risk Communication Advisory Committee to discuss methods and procedures to effectively convey and reduce risk to patients. The briefing and transcript information is available at www.fda.gov/ohrms/dockets/ac/08/transcripts/2008-4377t1-01.pdf. (Accessed July 19, 2010.)
S. On September 26, 2008, the FDA publically reported preliminary data from a German study in which an erythropoietin product not marketed in the U.S. (40,000 units daily for three days) and recombinant-tPA were used to treat acute ischemic stroke because there was an imbalance in the treatment arms for death. See www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm136211.htm. (Accessed July 19, 2010.)
T. On April 30, and May 1, 2009, the FDA convened a meeting of the Risk Communication Advisory Committee to discuss methods and procedures to effectively convey and reduce risk to patients. The briefing and transcript information is available at www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/RiskCommunicationAdvisoryCommittee/ucm158758.htm. (Accessed July 19, 2010.)
U. On April 30, 2009, the FDA revised the March 2007 boxed warning to address issues regarding ESA use by both patients with cancer and patients with chronic kidney failure.
- The warning noted that ESA dosing in oncology studies with hemoglobin targets of 12 g/dL or greater, whether the target was achieved or not, has resulted in more rapid cancer progression or shortened overall survival in cancer patients with advanced breast, head and neck, lymphoid and non-small cell lung malignancies and that these risks have not been excluded in cancer patients with hemoglobin targets of less than 12 g/dL
- The warning noted that ESAs should only be used to treat chemotherapy-induced anemia while patients are undergoing chemotherapy and not other types of anemia. (The indications section indicated that the chemotherapy should be myelosuppressive.)
- The warning noted that ESA dosing in renal disease studies with higher hemoglobin targets (e.g., 13.5 g/dL versus 11.3 g/dL and 14 g/dL and 10 g/dL), whether the target was achieved or not, has resulted in greater risks of death and serious cardiovascular events including heart attack, stroke and heart failure in pre-dialysis and dialysis patients. (In the non-boxed warning section, the warning noted an increased risk of mortality and cardiovascular complications in renal patients poorly responsive to ESA doses and given high ESA doses [CHOIR and NCHT trials cited.]).
V. In December 2009, the FDA issued the final version of the guidance for patient-report outcome measures. See www.fda.gov/downloads/Drugs/GuidanceCompliance RegulatoryInformation/Guidances/UCM193282.pdf. (Accessed July 19, 2010.)
W. In February 2010, the FDA required all ESAs to be prescribed and used under a risk evaluation and mitigation strategy (REMS) to ensure the safe use of these drugs. As part of the REMS, a Medication Guide explaining the risks and benefits of ESAs must be provided to all patients receiving ESAs. Information is available at www.fda.gov/AboutFDA/CentersOffices /CDER/ucm200847.htm, www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm109375.htm, www.fda.gov/AboutFDA/CentersOffices/ CDER/ucm200847.htm, www.accessdata.fda.gov/drugsatfda_docs/appletter/2010 /103951s5197ltr.pdf, www.accessdata.fda.gov/drugsatfda_docs/appletter/2010/103234s5199ltr.pdf. (Accessed July 19, 2010)
X. On October 18, 2010, the FDA convened a meeting of the Cardio-Renal Drugs Advisory Committee (CRDAC) to discuss safety issues for ESAs in TREAT trial. The briefing information is available at www.fda.gov/downloads/AdvisoryCommittees/.../Drugs/.../UCM236323.pdf. The transcript for the meeting is available at www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM233461.pdf. (Accessed July 19, 2010.)
Prior to the CRDAC meeting, Amgen submitted proposed labeling changes to the FDA regarding the use of ESAs in chronic renal failure patients not on dialysis that would limit treatment to patients who are most likely to benefit, specifically those with significant anemia (< 10 grams per deciliter ["g/dL"), and who are at high risk for transfusion and for whom transfusion avoidance is considered clinically important, including those in whom it is important to preserve kidney transplant eligibility. A more conservative dosing algorithm in these patients was also proposed. The sponsor also recommended against increased dosing in hyporesponsive patients. (See pages 88 and 89 www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM229328.pdf.) (Edgar 10-Q 08/09/10); accessed November 3, 2010)
VI. General Methodological Principles
When making national coverage determinations under section 1862(a)(1)(A) of the Act, CMS generally evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. The critical appraisal of the evidence enables us to determine to what degree we are confident that: 1) the specific assessment question(s) can be answered conclusively; and 2) the intervention will improve health outcomes for beneficiaries. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary.
A detailed account of the methodological principles of study design that the Agency utilizes to assess the relevant literature on a therapeutic or diagnostic item or service for specific conditions can be found in Appendix A. In general, features of clinical studies that improve studies and decrease bias include the selection of a clinically relevant cohort, the consistent use of a single good reference standard, and the blinding of readers to both the index test and the reference test results.
Public commenters sometimes cite the published clinical evidence and provide CMS with useful information. Public comments that provide information based on unpublished evidence, such as the results of individual practitioners or patients, are less rigorous and, therefore, less useful for making a coverage determination. CMS uses the initial comment period to inform the public of its proposed decision. CMS responds in detail to the public comments that were received in response to the proposed decision when it issues the final decision memorandum.
VII. Evidence
A. Introduction
We are providing a summary of the evidence that we considered during our review.
Emerging data have better delineated the physiologic criteria for intervention in the setting of anemia. Emerging data also suggest that ESAs are associated with increased mortality and morbidity despite the alleviation of anemia. The evidence reviewed in a prior NCD focused on ESA use in the cancer setting and related safety considerations. (www.cms.gov/medicare-coverage-database/details/nca-details.aspx?NCAId=203&ver=12&NcaName=Erythropoiesis+Stimulating+Agents+(ESAs)+for+non-renal+disease+indications&bc=BEAAAAAAAAAA&; accessed February 14, 2011.) The evidence reviewed in this NCA includes the literature on ESA therapy in populations with renal dysfunction, putative clinical benefits, and related safety issues. Studies were evaluated for information regarding dosage level, dose response, hemoglobin level, hemoglobin response, and correlation with clinical outcome(s). Studies comparing different ESA compounds or different routes of administration were included. The evidence reviewed encompassed studies germane to both dialysis and pre-dialysis patient populations. Materials found in published medical journal article were supplemented by data from additional technical sources as necessary.
B. Discussion of Evidence Reviewed
1. Question(s)
A. Is the evidence sufficient to conclude that the underlying cause for anemia in Medicare beneficiaries who have renal disease and are not on dialysis is absolute and irreversible erythropoietin deficiency?
B. If the answer to question A is affirmative, is the evidence sufficient to conclude that erythropoiesis (erythrocyte) stimulating agent (ESA) therapy affects health outcomes (including survival, cardiovascular event rates, exercise capacity, progression of renal disease, quality-of-life, transfusion rates, and ability to receive a transplant) when used by Medicare beneficiaries who have renal disease and are not on dialysis?
C. If the answer to Question B is affirmative, is there sufficient evidence to determine which characteristics of the patient, the disease, or the treatment regimen reliably predict a favorable or unfavorable health outcome when used by Medicare beneficiaries who have renal disease and are not on dialysis?
D. Is the evidence sufficient to conclude that the underlying cause for anemia in Medicare beneficiaries who have renal disease and are on dialysis is absolute and irreversible erythropoietin deficiency?
E. If the answer to question D is affirmative, is the evidence sufficient to conclude that erythropoiesis (erythrocyte) stimulating agent (ESA) therapy affects health outcomes (including survival, cardiovascular event rates, exercise capacity, quality of life, transfusion rates, and ability to receive a transplant) when used by Medicare beneficiaries who have renal disease and are on dialysis?
F. If the answer to Question E is affirmative, is there sufficient evidence to determine which characteristics of the patient, the disease, or the treatment regimen reliably predict a favorable or unfavorable health outcome when used by Medicare beneficiaries who have renal disease and are on dialysis?
2. External Technology Assessments
CMS requested two external technology assessments (TAs) on issues related to this technology.
The first technology assessment addressed changes in ESA utilization in the renal population. It was presented at the March 24, 2010 MEDCAC.(See Acumen slide set; www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/id78TA.pdf; accessed July 19, 2010.)
The second technology assessment addressed the impact of transfusions on renal transplant outcomes. The data were presented at the January 19, 2011 MEDCAC. (www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/id78TA.pdf; accessed February 2, 2011).
3. Internal Technology Assessment
a. Literature Search Methods
The reviewed evidence was gathered from articles submitted by the requestor and a search of the published literature, government databases, and other online references. CMS staff extensively searched Medline (1988 to present) for primary studies evaluating ESA therapy in renal disease. The emphasis was on studies structured to assess long-term health outcomes with hard clinical endpoints. CMS staff likewise searched for systematic reviews and technology assessments from other sources such as the Cochrane collection and the Agency for Healthcare Research and Quality (AHRQ) library. Systematic reviews were used to help locate some of the more obscure publications and abstracts. For material outside the domain of the published medical literature, additional sources were used.
CMS reviewed FDA reviews of the registration trials for erythropoietin alpha, darbepoetin alpha, and methoxy polyethylene glycol epoetin beta, as well as the FDA safety data for the two marked compounds, erythropoietin alpha and darbepoetin alpha. CMS also reviewed published data on other erythropoiesis stimulating agents not marketed in the U.S. CMS reviewed the transcripts and briefing documents (FDA and pharmaceutical sponsor) from the 2004 FDA Oncologic Drug Advisory Committee meeting, the 2007 FDA Cardio-Renal Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee meeting, and the 2010 FDA Cardio-Renal Drugs Advisory Committee meeting on ESA safety. CMS reviewed the FDA ESA drug safety alerts and label changes. CMS reviewed the development of the risk evaluation and mitigation strategy (REMS) program for ESAs. CMS searched the National Institutes of Health Clinical Trials.gov database for ongoing/completed trials of ESAs. CMS used internet searches to identify websites with clinical trial results, press releases for clinical trial termination, and U.S. government regulatory action. Preference was given to English publications, phase III and IV randomized, controlled studies with hard clinical endpoints (vs pilot studies or dose ranging studies), studies involving adults, and ESAs approved for use in the U.S.
Keywords used in the searches included: anemia and physiology, renal, kidney, dialysis, or pre-dialysis, chronic kidney disease (CKD), or end stage renal disease (ESRD); ESAs (erythropoietic stimulating agents, erythropoiesis stimulating agents, erythropoietin, epoetin, darbepoetin, pegylated erythropoietin, erythropoietin receptor activator, CERA, continuous erythropoietin receptor activator, peginesatide, hematide, or mimetibody) and anemia, dosing, pharmacokinetic-pharmacodynamic (PK-PD), transfusion, renal disease progression, exercise, (health-related) quality-of-life, pure red cell aplasia (PRCA), thrombosis, cardiovascular, tumor progression, morbidity, survival, mortality, renal transplantation, or resistance; transfusion and anemia, physiology, risk, renal transplantation, sensitization, panel reactive antibodies (PRA), or HLA-specific antibodies; renal transplantation and demographics, surgical criteria, UNOS data collection, immune suppression, protocols for sensitized patients, panel reactive antibodies (PRA), or HLA-specific antibodies; panel reactive antibodies (PRA) and HLA specific antibodies, assay type, or risk factors.
b. Evidence Review Findings
Summary
Despite an exhaustive search, we identified no high quality, randomized clinical trials that were of sufficient design, duration, and power to confidently conclude that ESAs provide clinical benefits other than increasing hemoglobin, a putative intermediate clinical surrogate. Despite an exhaustive search we identified no high quality, randomized clinical trials that were of sufficient design, duration, and power to definitely determine the absolute risk of adverse events including death, tumor progression, and cardiovascular-thromboembolic events in patients with renal insufficiency and/or renal failure, in geriatric patients (the largest growing renal population segment), using ESAs. No trials were structured to assess these hard endpoints stratifying by renal disease severity (and stage ascertained by studies other than estimated GFR), by entry hemoglobin in ESA-naïve patients, by prior ESA response, by ESA response after a limited number of doses, by a priori bone marrow reserve documented by biopsy studies, by concomitant drugs such as angiotensin converting enzyme inhibitors, by age, and by various co-morbidities. No trials eliminated a) the confounding associated with hemoglobin levels and targets and b) effects that might be non-linear by randomizing blinded cohorts with fixed dosing. No trials were structured to assess transfusion endpoints (number units, number persons, frequency, transfusion reason, antecedent hemoglobin) with a priori transfusion criteria based on accepted data-based criteria for transfusion. No trials used appropriately validated health-related quality-of-life (hrQOL) instruments and established clinically significant differences related to hemoglobin levels and change in hemoglobin levels. No trials limited dosing to physiologic replacement. No trials were structured to assess hard clinical outcomes in settings in which the ESA level is supra-physiologic because of dose itself, drug plasma-clearance/tissue residence times, the route of administration, or the dosing interval. No studies were adequately structured assess within class safety differences for ESAs. We did identify 4 large, randomized studies that were structured to assess survival or cardiovascular endpoints (Besarab 1998, Drueke 2006, Singh 2006, Pfeffer 2009). All used hemoglobin targets and none used fixed ESA dosing. Only one was placebo controlled. None included many of the types of patients that have become more common in the CKD population. Two were terminated early. High withdrawal rates complicated many of the studies. We did identify unpublished studies submitted to the FDA for registration and multiple studies which compared routes of administration, different treatment regimens, or different ESA agents. We detail our findings below.
i. Hypothesis Generating Studies
Although physiologic dysfunction with renal disease is multi-factorial, it was postulated that anemia might play an important role in exercise capacity, rate of renal function decline, cardiac morphology, and survival.
A cross-sectional study of 13 dialysis patients (Hb range 5.1-12.2 g/dl) by Mayer et al (1989) demonstrated that the impairment in O2 uptake at the anaerobic threshold was inversely related to the hemoglobin level. Maximum peripheral O2 uptake was similarly correlated with hemoglobin levels.
Three randomized studies estimated the rate of decline in kidney function using surrogate measures. Kuriyama et al. reported that serum creatinine doubled in 26/31 (84%) anemic pre-dialysis patients not treated with erythropoietin versus 21/35 (60%) of non-anemic pre-dialysis patients not treated with erythropoietin versus 22/42 (52%) anemic pre-dialysis patients treated with erythropoietin for 36 weeks and followed for a median duration of 28 months. (Kuriyama 1997) (The differences between groups 2 and 3 were not statistically significant.) Limited data suggested that the presence of diabetes might reduce the effect of erythropoietin on progression. A study by Teplan et al. (n = 186) using inulin clearance changes suggested that supplementary dietary ketoacids and erythropoietin might independently contribute to decreased progression in patients on a low protein diet.(Teplan 2001a,b, Teplan 2003) Gouva et al. reported that the composite endpoint of serum creatinine doubling, initiation of dialysis, or death was met in 23/43 (54%) of those in whom erythropoietin treatment was delayed until hemoglobin levels decreased to less than 9 g/dl as compared 13/45 (29%) of those in whom treatment was initiated for milder anemia (hemoglobin 9 to 11.6 g/dl). (Gouva 2004)
A cross-sectional study of 78 dialysis patients by Silverberg et al. (1989) demonstrated that left ventricular mass was inversely related to hemoglobin levels (slope = [-1.2 g/m2]/g/l hb): quartile 1 (hb < 7.7 g/dl) 158 ±6 g/m2, quartile 2 (hb 7.7-8.8 g/dl) 140±10 g/m2, quartile 3 (8.8 -9.7 g/dl) 132 + 7 g/m2, and quartile 4 (hb > 9.7 g/dl) 120+8 g/m2 (and positively correlated to even modest systolic blood pressure elevation [slope = [0.57 mg/m2]/mm Hg]).
An observational study data conducted by Ma et al. (1999) using USRDS data reported that all-cause and cardiac death rates were highest in patients with the lowest hematocrit levels (Table 2). (Collins 1997, 2000, 2001, 2002, Ma 1999) Patients with diabetes had higher rates of both all-cause and cardiac than did non-diabetic patients. (No distinctions were made for type 1 vs type 2 diabetes.) (See Analysis.)
Table 2: Mortality and Anemia: Observational Data from USRDS
It was not known whether anemia management and therapeutic intervention with ESAs (and other agents) would improve the physiologic dysfunction associated with renal disease. At the time that ESAs were being developed, there were concerns about the use of transfusions and the safety of the blood supply (HIV and non-A/B hepatitis).
ii. Initial Pivotal Registration Studies
Erythropoietin-alpha (Trade names: Epogen and Procrit) was approved an orphan drug (< 200,000 patients) for use in renal patients in 1989 (Asbury 1991, Coster 1992, FDA Summary Basis of Approval for BLA # 103234, NKF Position Paper 1989, Phase IV commitment study Nissenson 1991). Only three of the major registration studies have been published in full: 1) a blinded study of hemodialysis patients (Canadian Study Group) (86-004), 2) an uncontrolled study in hemodialysis patients (Eschbach) (8601), and 3) a blinded study of pre-dialysis patients (Teehan)(G88-011) (Table 3, Panels A, B, and C). Some of these studies were also presented as sub-studies or ancillary studies. Other registration studies were not published or were only sub-studies published by individual investigators. Multiple citations delineated in early product labels could not be located. The FDA reviews of the registration studies are not available.
Table 3A: FDA Registration studies-Erythropoietin alpha*
Table 3B: FDA Registration studies-Erythropoietin alpha (continued)*
Table 3C: FDA Registration studies-Erythropoietin alpha (continued)*
The registration clinical trials for erythropoietin-alpha assessed patient populations that differ from current renal populations. Many of the subjects were substantially more anemic than subjects in later trials. The mean hemoglobin in the Canadian study of hemodialysis patients was less than 7g/dl. Many of the subjects were substantially younger. The age in the Canadian study of hemodialysis patients was approximately 15 years younger than current hemodialysis patients. (Canadian Group, USRDS 2008, 2009) The Canadian study excluded patients with many co-morbidities including type 1 diabetes and patients who would not be likely to complete the exercise testing. Incident rates for diabetes in the dialysis population have doubled since 1990 (although the USRD data do not distinguish between type 1 and type 2 diabetes). (USRDS 2008, 2009) More than 36% of current dialysis patients have walking disabilities and more than 26% use assistive devices. (USRDS 2008) Co-morbidities markedly increase the likelihood of wheelchair use. (USRDS 2008)
The registration trials for erythropoietin-alpha did not distinguish between the various stages of pre-dialysis renal disease and used an insensitive measure of glomerular filtration function, (serum creatinine 3-10 g/dL). Causes of anemia other than iron, folate, and B-12 were not excluded. Bone marrow biopsies were not obtained. Multiple myeloma was indentified incidentally in one patient.
The registration trials did not always account for all patients or conduct intent-to-treat analyses. Amgen briefing materials indicate that 426 patients entered the single-arm phase III 12+ week trial (www.amgen.com/pdfs/misc/2007-AMGEN-FDA-CADRC.pdf; accessed July 19, 2010). Published materials suggest that only 333 patients entered the study (Eschbach 1989) and that only 309 had evaluable data (Adamson 1989). Reportedly only 266 remained on therapy 13 months after study initiation. The drop-out rate in the 6-month Canadian study was 16%. Subjects were not assessed unless they completed outcome assessments at four time points. There were no intent-to-treat analyses. The drop-out rate in the 8-week Teehan study was 10% and was due to adverse events. (US Recombinant Human Erythropoietin Pre-dialysis Study Group [Teehan] 1991) Curiously most of the drop-outs in the placebo cohort occurred early (10.5 days) versus late in the treatment cohorts (36.0 days). The presence of cancer in three participants raises questions about the screening procedures or tumor promotion (See TREAT study). The statistical plan did not delineate whether per-protocol or intent-to-treat analyses were conducted.
The registration trials were relatively small, short in duration, and focused on surrogate endpoints (hemoglobin [hematocrit] levels and changes in hemoglobin [hematocrit] levels), transfusion reduction, and quality-of-life including self reports of physical function (Tables 4 and 5). Hemoglobin levels did increase for many patients, but the studies provided no information on the characteristics of patients who required more than physiologic replacement to obtain a response or who did not respond. Nor did the studies provide information on the likelihood of response based on the pre-treatment hemoglobin (hematocrit) level. No patients were transfused in the pre-dialysis study (Table 5). Twenty five patients were transfused in the hemodialysis study and most of these were in the placebo arm (Table 4). There was an imbalance at baseline for transfusion dependence in favor of the high target erythropoietin arm. There were, however, no validated hemoglobin (hematocrit) thresholds for initiating transfusion. Nor were there pre-specified transfusion protocols. Information on the number of units transfused, the number of units per transfused person, the reason for transfusion, and the characteristics of the patients who received transfusion was lacking.
Quality-of-life data were submitted for the published Canadian hemodialysis (86-004) and the uncontrolled open-label 8601 studies. Reportedly data were also submitted for two unpublished studies in hemo- and peritoneal dialysis patients (8701 and 8904). None of the instruments used were validated to assess health-related quality-of-life in the populations studies. Some studies employed modified instruments and post-hoc analyses. There were no pre-specified power calculations based on values and changes in values established to be clinically meaningful. There were no pre-specified plans for addressing missing data. Changes in anemia symptoms and health-related quality-of- life parameters did not correlate with hemoglobin levels and changes in hemoglobin levels (±stratification based on baseline hemoglobin levels). The open-label design limited any interpretation of the self-report data. The short study lengths did not permit assessment of durability of any health-related quality-of-life improvements potentially attributable to a drug intended to be given on a chronic basis. The exclusion criteria for co-morbid conditions did not permit assessment of any health-related quality-of-life improvements in sicker populations. Although such claims were initially present in the label (…Once the target hematocrit (32% to 38%) was achieved, statistically significant improvements were demonstrated for most quality of life parameters measured, including energy and activity level, functional ability, sleep and eating behavior, health status, satisfaction with health, sex life, well-being, psychological effect, life satisfaction, and happiness. Patients also reported improvement in their disease symptoms. They showed a statistically significant increase in exercise capacity (VO2 max), energy, and strength with a significant reduction in aching, dizziness, anxiety, shortness of breath, muscle weakness, and leg cramps…), after re-analysis by the FDA, the claims were removed the label and the FDA issued a guidance document for patient- reported outcome (PRO) claims. (2009 FDA Guidance Document for Patient-Reported Outcomes, Trentacosti 2007 Slide Set)
Table 4: Anemia and Transfusion in the Canadian Group Study: Hemodialysis 6 Month Study (Mean Age Mid 40s)
Table 5: Anemia and Transfusion in US Human Recombinant Erythropoietin Pre-dialysis Study Group (Teehan 1991) 8 Week Study (Mean Age 57.1 yrs)
Although hypertension and thrombosis were observed, the registration studies were not structured to assess mortality, chronic morbidity, and less frequent adverse events. Although reversible bone marrow fibrosis, which would be distinct from that associated with profound hyperparathyroidism in some dialysis patients, was observed in the longer rodent and canine studies, no large and long-term studies with randomization (or stratification) by ESA dose assessed bone marrow changes.(Akada 2010, Bader 1992, Barosi 2005, Dokal 1989, Epogen label, Gallieni 2000, Kakumitsu 2005, Lacout 2006, Levine 2005, Reilly 1997, Tulliez 1989, Wernig 2006) Although animal carcinogenicity studies are frequently required for drugs, including hormones which can act as growth factors, e.g., insulin products, there were no such studies in the registration package. None of the registration clinical trials were long or large enough and included the appropriate patient populations to exclude oncogenic or promoter activities—especially with supraphysiologic doses (either via compressed dosing regimens, intravenous route of administration, or dose levels). Drug exposure in the registration trials was insufficient to reveal the subsequently identified antibody-mediated red cell aplasia associated with either long-term exposure to the active agent or package leachates. (Boven 2005, Howman 2007, Jacob 2006, Ryan 2006, Schellekens 2006) The registration studies for erythropoietin did not include analysis of safety and efficacy in geriatric patients (≥65 years) and racial-ethnic groups. Nor did they include drug interaction studies-although medications frequently used in the renal population, e.g., the anti-hypertensive, anti-protienuric angiotensin-converting enzyme (ACE) inhibitors are thought to impair erythropoietin (endogenous and exogenous) efficacy. (Cruz 1996, Hayashi 2001, LeMeur 2001, Onoyama 1989, Quereshi 2007, Ripamonti 2006)
ii. Pivotal Registration Studies for Analogues
aa. Darbepoetin (Trade name: Aranesp)
The pivotal registration trials for darbepoietin were non-inferiority studies (Table 6). (Nissenson 2002, Varenterghem 2002) They included only patients who had previously been on ESAs. The populations were different than the original erythropoietin populations. In double-blind Study (970)117 based in North America, the 522 hemodialysis patients were more than a decade older (mean 57.9 years, range 20-90 years), they were less anemic albeit not ESA-naive, (mean hemoglobin 11.2 g/dl; range 9.6-12.6 g/dl), and hypertension and diabetes were found in 26% and 35%. The mean erythopoietin dose at entry was 13,776 U/week (range 1200-120,000). (Weekly dose for a 70 kg person dosed at 50 U/kg is 10,500 units.)
In open-label study (970)200 based in Europe and Australia, the 522 dialysis patients were more than a decade older (mean 60.4 years, range 18-88 years), they were less anemic, (mean hemoglobin 11.0 g/dl; range 9.5-12.5 g/dl), and hypertension and diabetes were found in 8% and 15%. The median erythropoietin dose at entry was 6,000 U/week (quartiles 4,000-9,000) (half of the 117 entry dosing). The randomization for darbepoetin:erythropoietin was 1:2 for study 117 (reportedly an error, but one which limited darbepoetin exposure) and 2:1 for study 200.
Neither study used fixed doses. Study 117 used only IV administration whereas Study 200 used both SQ and IV administration. Although the studies excluded patients with more established risk factors for ESA resistance such as inflammation, neither study assessed the potential impact of ACE inhibitors or ARBs on efficacy. Neither study had an algorithm for transfusion use nor did neither report transfusion results (Table 9). Non-compliance and drop-out was high, limiting per-protocol analysis to approximately 70% of the initial population. For study 117, the death rates during the study or the 30 day follow-up period after last dose were 5% (9/169) for the darbepoetin arm and 7% (23/338) for the erythropoietin arm. For study 200, the death rates during the study, by the last contact date, and/or the 28 day follow-up period after the last dose were 12% (41/346) for darbepoetin and 6% (11/173) for erythropoietin (p = 0.06). Reportedly, the death rates converged at two year follow-up (19% vs 17%). Although these data suggest different time-to-death profiles for the two ESAs, survival curves were not provided. There was no analysis and discussion of the role that the different study doses might have played in the different mortality outcomes.
Two other major clinical studies were included in the registration package (Unpublished Study 211, Locatelli 2001 Study 980202. See ESA Type). Study 202 was open-label and enrolled 166 ESA-naïve, pre-dialysis patients for 3:1 darbepoetin:erythropoietin randomization with doses to be titrated over 24 weeks. Study 211 open-label and enrolled 122 ESA-naïve dialysis patients for 3:1 darbepoetin:erythropoietin randomization with doses to be titrated over 20 weeks. In both studies the major contributing causes to renal disease were diabetes and/or hypertension. The pre-dialysis patients were almost 8 years older than the dialysis patients. Both populations were less anemic than the original erythropoietin populations: Study 211 basal hemoglobin 8.6 g/dl; Study 202 basal hemoglobin 9.4 g/dl. Neither study was designed for rigorous statistical evaluation as either superiority or non-inferiority trials. The results are most notable for high frequency of transfusion in the darbepoetin arm, 27% of patients, versus the erythropoietin arm, 16% of patients. This study remains unpublished.
The registration package did not include drug interaction studies, animal/human marrow studies for fibrosis (and resistance), and animal carcinogenicity studies.
The FDA review concluded that darbepoetin and erythropoietin are equivalent ESAs. Darbepoietin, however, does not carry the indication for transfusion reduction (only anemia management) because non-inferiority designs were used in the pivotal registration studies. In addition, they noted that the pharmacokinetic relationship between the compounds is not linear and that IV administration may require higher dosing than with SQ administration (Table 9). Their composite analysis of the registration studies reportedly demonstrated equivalent safety and efficacy in geriatric patients (486 patients aged 65 to 74 years and 306 patients aged 75 years and older). There were 360 non-Caucasian patients (Black n = 234, Asian n = 54, Hispanic n = 36, Other = 36) in the study populations; limiting conclusions about safety and efficacy in racial-ethnic groups. The absence of placebos control and fixed doses in the clinical studies limited the conclusions that could be drawn about compound specific effects versus ESA class effects and the role of hemoglobin level versus dose on safety endpoints.
Table 6A: FDA Registration studies-darbepoetin alpha
Table 6B: FDA Registration studies-darbepoetin alpha (continued)
Table 6C: FDA Registration studies-Darbepoetin alpha (continued)
bb. Pegzerepoetin (Trade name: Mircera)
The six pivotal registration trials for methoxy polyethylene glycol-epoetin beta (pegylated erythropoietin) were non-inferiority studies (Table 7). (Canaud 2008, Klinger 2007, Levin 2007, Macdougall 2008, Spinowitz 2008, Sulowicz 2007) None of the studies were open-label. None had algorithms for transfusion use (Table 9). All excluded patients with inflammatory conditions that might induce ESA resistance.
Although the FDA medical officer review reported the inclusion of 559 patients 65 to 74 years of age (22%) and 508 patients 75 years of age or older (20%) in the pivotal trials, the label stated that there were insufficient numbers of patients for analysis of efficacy and safety in the geriatric population. The review also reported the inclusion of 476 patients of African descent (19%) and 127 patients of Asian descent (5%) in the pivotal trials. The FDA reviewer did note a higher incidence of death in Asian patients exposed to pegzerepoetin (5%) than Asians in the reference arms (2%), but cautioned about over-interpretation.
The registration package did not include animal carcinogenicity studies, animal/human marrow studies for fibrosis (and resistance), or drug interaction studies. The FDA review did note that more patients in the pegylated erythropoietin treatment arms (7.5%) than patients in active control ESA arms (4.4%) were likely to have decreased platelet counts (< 100x109/L) and that there were more patients with serious bleeding episodes (and gastrointestinal hemorrhage in particular) in the pegylated arms (5.2% [1.2 %]) versus the ESA reference arms (4% [0.2 %]). The report did not provide any correlative information about these adverse events: whether the thrombocytopenia was related to the serious bleeding or whether the thrombocytopenia was related to marrow fibrosis or poor marrow reserve in the setting of chronic supraphysiologic ESA stimulation.
The FDA review concluded that pegylated erythropoietin-beta is equivalent to the other approved ESA, darbepoetin and erythropoietin-alpha. Pegylated erythropoietin, however, does not carry the indication for transfusion reduction (only anemia management) in renal disease because non-inferiority designs were used in the pivotal registration studies (Table 9). (Pegylated erythropoietin-beta is not indicated for anemia in the oncologic setting; drug development for this indication was terminated because of increased mortality in an early comparative dose ranging study.) The absence of placebos control and fixed doses in the clinical studies limited the conclusions that could be drawn about compound specific effects versus ESA class effects and the role of hemoglobin level versus dose on safety endpoints.
Table 7A: FDA Registration studies-Pegylated erythropoietin-beta
Table 7B: FDA Registration studies-Pegylated erythropoietin-beta (continued)
Table 7C: FDA Registration studies-Pegylated erythropoietin-beta (continued)
cc. Peginisatide
There were four pivotal trials intended for registration of the long-acting erythropoietin receptor stimulator, peginisatide (formerly known as hematide). All utilized an open-label, non-inferiority design (Table 8) (Analyst Day handout) Two were conducted in pre-dialysis patients (PEARL 1 and 2); two in dialysis patients (EMERALD 1 and 2). Hemoglobin changes from week 29 to 36 weeks (primary endpoint), the percentage of patients with hemoglobin increases > 1 g/dl and hemoglobin > 11 g/dl from week 29 to 36 weeks (secondary endpoint), and the percentage of patients who transfused during the 36 week study (secondary endpoint) were equivalent to predicate ESAs in dialysis populations. (These endpoints, however differed from those delineated in ClinicalTrials.gov and listed in Table 8C) (www.finance.yahoo.com /news/Affymax-to-Webcast-Analyst-bw-910437963.html?x=0&.v=1&vm=r; accessed November 10, 2010; www.shareholder.com/visitors/event/build2/mediapresentation.cfm?companyid=AFFY&mediaid=45251&mediauserid=4919438&TID=1078036874:aa4491b89ab2533a970727d26a7a8006&popupcheck=0&shexp=201102071258&shkey=71daf8baad92c9d1eb8eab268072410d&player=; accessed November 29, 2010; Piper Jaffray Healthcare Conference webcastingplayer.corporate-ir.net/player/PlayerHost.aspx?EventId=3497574&Stream Id=1599057&TIK={B08FA7B7-20ED-4444-83F5-92B77BAF8ACB}&RGS=1; accessed December 1, 2010; www.talkpoint.com/content/17720C7F-49B7-4601-9993-DF7181F618CB/EE00B7DE-7621-4FBC-BC79-37F2B1B47529/35B0C560-15DB-48DB-B55F-AD3F0786CC5B/3/AffymaxAnalystDay122.pdf; accessed December 2, 2010.)
In the PEARL 2 study, more patients on low and high dose peginesitide, 11.4% and 10.4%, versus 4.9% on darbepoetin received transfusions. There were similar trends, although less robust, in PEARL 1. There were more patients with cardiovascular events (death, stroke, myocardial infarction, congestive heart failure, unstable angina, and arrhythmia) in the pooled PEARL studies: 21.6 % in the peginesatide arm versus 17.1% in the erythropoietin arm. The largest differences were seen in death (8.8% versus 6.7%, arrhythmia 2.4% versus 4.0%, and unstable angina 2.4% versus 0.9%). Most of the differences were found in PEARL 2; some, but not all were attributed to baseline imbalance.
Table 8A: FDA Registration studies-Peginesatide
Table 8B: FDA Registration studies-Peginesatide (continued)
Table 8C: FDA Registration studies-Peginesatide (continued)
Table 9: Anemia and Transfusion in ESA Analogue/Receptor Activator Pivotal Trials
iii. Other Potential Benefits from ESAs
We looked for other potential benefits from erythropoiesis stimulating agents including exercise capacity for activities of daily living, intermediate surrogates for cardiac function, progression to dialysis, and health-related quality-of-life measures.
aa. Exercise Capacity (Endurance; Strength)
We identified eight randomized studies with ESA as a treatment arm and objective measures of exercise capacity as endpoints (Table 10). Studies with patient-reported (n = 22) or physician-reported assessment (n = 2) of physical function were not included.
One of these studies (Furuland 2003), however, changed its focus from exercise to safety when many of the recruited subjects were unable to complete exercise testing. Another one of studies (Palazzuoli 2006) was conducted in congestive heart failure patients with some renal insufficiency and anemia. The congestive heart failure inclusion criteria were well defined and characterized: New York Heart Association Class 3 or 4 whereas the renal criteria were less well defined: serum creatinine less than 5 mg/dl (actual: 2.4 ±0.5 g/dl).
Of the seven studies with exercise results, six were conducted in adults. One was conducted in children. Six were nominally double-blind. The largest study by Parfrey et al. blinded the patients and those conducting the assessment, but not the treating physicians. Of the remaining two studies, one was single-blind and the other open-label. Six studies were conducted in patients on dialysis; two were conducted in the pre-dialysis patient population. Four of the studies compared ESA treatment to no ESA treatment; one of these also employed hemoglobin target level cohorts. One of the studies included an exercise training variable in addition to ESA treatment at two hemoglobin target levels. Two studies had more than 100 participants. The first with n = 596 had a 54% completion rate; the other with n = 118 had an 84% completion rate.(Canadian 1990, Laupacis 1990, 1991, Parfrey 2005) Only one study, by Parfrey et al., had treatment arms longer than 12 months in duration. Many of the studies assessed peak oxygen consumption (VO2max). Others assessed time or distance walked/biked-often, but not always, with formal stress testing. The baseline imbalance for exercise capacity in two studies was not addressed. (Canadian 1990, Clyne 1992, Laupacis 1990, 1991)
The studies reveal no consistent improvement in exercise capacity. In the largest study by Parfrey et al., there was intra-group improvement in the six-minute walk test for both of the treatment arms among patients who completed the study although there was no inter-group difference. There was no intra-group improvement for either treatment group when intent-to-treat analyses with last observation carried forward were conducted. In other words, there were no improvements when available results from the drop-out population (46%) were included-suggesting differences between the completer and drop-out patient populations regardless of treatment cohort. Even in the studies with reported improvement, performance results were noted to be sub-optimal. (McMahon 1999, 2000, Painter 2002) Analyses evaluating any potential correlation between hemoglobin and exercise capacity or between the change in hemoglobin and the change exercise capacity were not performed except in the Palazzuoli et al. study in congestive heart failure patients with mild renal insufficiency (N = 38). (Palazzuoli 2006) Indeed in the Painter et al. study with its four treatment arms, VO2 improved in both of the treatment arms with exercise training regardless of hemoglobin target. A higher hemoglobin target did not confer any benefit for functional capacity.
Table 10: Exercise Studies
Of note, Leikis et al. followed a small cohort of 12 patients with stage 3-4 chronic renal insufficiency with exercise performance testing (fatigue with isokinetic dynamometry, leg extension strength, peak VO2) and observed deterioration in exercise function in concert with renal decline function despite maintenance of hemoglobin levels. (Leikis 2006) These data suggested the importance of factors other than hemoglobin in exercise capacity.
Three related studies also suggested benefit from exercise training itself. Kouidi et al. studied seven hemodialysis patients before and after a 6-month thrice weekly exercise program including stretching, resistance, and aerobic activities. (Kouidi 1998) The mean hematocrit did not change during the study 10.9 to 10.4 volume %. Exercise duration (29%) and peak VO2 (48%) improved. Lactate levels (16%) decreased. Although morphologic evidence of atrophy persisted, concomitant muscle biopsies showed an increase in muscle volume: type 1 fibers (slow twitch) (26%) and type 2 fibers (fast twitch) (24%).
De Paul et al. randomized 38 hemodialysis patients into two open-label exercise programs: resistive isotonic quadriceps/hamstring strengthening and endurance training on a cycle ergometer or a range-of-motion exercises for 12 weeks.(DePaul 2002) Erythropoietin use, hemoglobin levels (11.6 vs 11.1 g/dl), and dialysis adequacy were similar for the two groups. Exercise sessions were conducted at the time of dialysis. Maximal ergonomic workload changed from 21 to 44 watts in the strengthening/endurance training group and from 22 to 30 watts in the range-of- motion exercise group. Thigh strength changed from 166 to 228 lb in the strengthening/endurance training group and from 171 to 173 in the range-of-motion exercise group. A distance walked in a six minute interval changed only 460 to 464 meters in the strengthening/endurance training group and from 426 to 430 meters in the range-of-motion exercise group. Curiously, the mean SF-36 and Kidney Disease Questionnaire scores did not change by treatment group. The nine patients who did not complete the exercise assessments reportedly had worse baseline physical functioning at baseline and more co-morbidity. Although the exercise programs may have contributed to improvements in strength, they did not normalize function.
In a similar study, Ouzouni et al. randomized 35 patients to an exercise program or a no-exercise treatment arm. Exercise sessions were conducted at the time of dialysis. The exercise program consisted initially of 30 minutes each of cycling and strengthening/flexibility exercises. (Ouzouni 2009) Duration and workload were increased over time. Among the 33 subjects who completed the trial, the duration of exercise during a modified Bruce protocol treadmill test changed from 16.9 to 20.9 minutes in the exercise arm and 15.9 to 15.1 minutes in the placebo arm. Exercise capacity changed from 9.1 to 11.2 metabolic equivalents of task (METs) and 8.7 to 8.9 METs in the placebo group. Peak VO2 changed from 20.9 to 25.3 ml/kg/min in the exercise arm and from 20.3 to 20.1 ml/kg/min in the control arm. Exercise, but not hemoglobin level, was identified as the contributory factor to improved quality-of-life scores in regression analyses.
A survey study by Kontos et al. identified barriers to exercise participation by older hemodialysis patients. (Kontos 2007)
bb. Intermediate Surrogates for Cardiac Outcomes
Left ventricular hypertrophy and poor cardiac output in renal patients have been linked with anemia and poor clinical outcomes. (Eckhardt 2001, London 1989, metivier 2000, Okada 1989, Silverberg 1989) We identified ten randomized studies with ESA as a treatment arm and objective measures of cardiac function as endpoints.
Table 11: Intermediate Cardiac Surrogate Studies
cc. Progression to Dialysis
As noted in the Hypothesis Generating section, we identified three pilot studies which reported improvements in the rate of renal function decline using surrogate measures. (Gouva 2004, Kuriyama 1997, Teplan 2001 a,b, Teplan 2003)
We also note four additional studies of renal decline using surrogate endpoints. Roth et al. studied changes in renal function over 48 weeks in 83 pre-dialysis patients treated with erythropoietin or placebo. (Roth 1994) The open-label study, which was performed to exclude a negative consequence of erythropoietin exposure, did not reveal any treatment related differences in GFR change (125I-iothalamate clearance).
Similarly, Kleinman et al., in what appears to be a subset of an unpublished, randomized registration study, followed reciprocal serum creatinine changes in eight of 14 patients over 12 weeks in an attempt to exclude secondary accelerated renal decline. (Kleinman 1989)
In a two year open-label study, Roger et al. assessed changes in left ventricular hypertrophy (LVH) by echocardiography (primary endpoint) and renal function by calculated creatinine clearance, 51 Cr-EDTA or 99mTc-diethylenetriamine penta-acetic acid clearance, or progression to dialysis (secondary endpoint) in 155 pre-dialysis patients randomized to hemoglobin targets of 12 to 13 g/dl versus 9 to 10 g/dl. (Roger 2004) Renal function testing reportedly did not differ by treatment group, but there was a trend (p = 0.08) to increased initiation of dialysis 24 (32%) in the high target arm versus 15 (19%) in the lower target arm.
The ECAP (Effect of Early Correction of Anemia on the Progression of CKD) open-label study by Rossert et al., but written by Dr. Amy Ferry (Medica Excerpta) with Ortho Biotech funding, had a primary endpoint of rate of GFR decline using plasma iohexol clearance, a planned enrollment of 630 subjects, and a scheduled duration of 40 months (four months of titration and stabilization and 36 months of maintenance). (Rossert 2006, 2007) The study, however, was terminated early reportedly because of emerging safety concerns about pure red cell aplasia (PRCA) with subcutaneous administration. (Boven 2005, Jacob 2006, Howman 2007, Ryan 2006, Schellekens 2006) (Indeed, two cases of occurred in the high target arm.) Enrollment in the two treatment arms (hemoglobin targets 14.0-15.0 g/dl for men and 13.0-14.0 g/dl for women versus 11.0-12.0) was limited to n = 391. Two-hundred forty-one subjects completed the stabilization phases and entered the maintenance phase for a mean follow-up of approximately eight months. Two or more GFR measurements were available for n = 163. Changes in GFR did not differ by treatment group and were substantially less than expected. The blunted progression was attributed to ACE inhibitors, blood pressure targets, and lipid control.
We identified three randomized studies which reported data on renal disease progression to end-stage renal disease, a more definitive endpoint (Tables 12 and 23). This endpoint was not the primary outcome parameter for any of the studies. All were multi-year studies and all had more than 500 patients. Two were open-label (CHOIR and CREATE); one was blinded (TREAT). Each study employed a different ESA. Baseline renal function data in all studies was limited by the use of serum creatinine and formulas to estimate glomerular filtration (GFR). No study conducted analyses correlating changes in hemoglobin (with or without stratification by baseline renal function and/or baseline [ESA naïve] hemoglobin) with changes in GFR. None of the studies showed that use of ESAs to achieve a higher hemoglobin target resulted in a decreased likelihood of progressing to end-stage renal disease and the need for dialysis. Indeed in the CREATE study, the difference between the treatment cohorts reached statistical significance. Comparative ESA dose information on those who progressed to end-stage renal disease and those who did not was not available.
Table 12: Studies of Progression to Dialysis
Of note, dialysis adequacy as measured by Kt/V ([Dialyzer Clearance of Urea x Dialysis Time]/Volume Urea Distribution) was not better after treatment in the higher hemoglobin target arm (1.35; change -0.03)(n = 618) versus in the lower hemoglobin target arm (1.44; change + 0.06) (n = 612) in the Normalization of Hematocrit Trial.(Besarab 1998, KDOQI Hemodialysis Adequacy Guidelines 2006) A higher proportion of patients in the higher target arm (32% ) had endpoint Kt/V values below 1.20, the minimal level for dialysis adequacy, compared to patients in the lower target arm (22%).
dd. Health-related Quality-of-Life
We identified 11 blinded, randomized studies which reported use of quality-of-life measures (Table 13). Studies which compared different treatment regimens, other than hemoglobin targets, were excluded. Two studies (8701 and 8904) submitted for the initial erythropoietin NDA submission and resubmitted for the 2007 FDA advisory committee meeting on ESAs and quality-of life-measures have never been published and were not available for review despite requests to the FDA and the sponsor (Amgen). (See FDA section.)
Most of the identified studies were small and of limited duration. None of the studies described employed instruments of health-related quality-of-life that were validated in the population to be studied.(2009 FDA Guidance to Industry on PRO Claims) None of the studies were powered a priori for health-related quality-of-life testing based on biologically significant changes. (In addition, because the sponsor declined to provide information about SF-36 survey, which is proprietary, it was not possible to determine the clinical relevance of specific score levels and changes in scores.) Some studies selected subsets of test instruments. Some studies tested at multiple time-points or used multiple instruments, but did not apply Bonferroni corrections for multiple measures. In studies in which several instruments were used, results were not internally consistent. Frequently testing and analysis occurred only in completer populations. Because many of these studies had high drop-out rates, results cannot be applied to the enrolled patient populations or extrapolated to the general renal population. Putative improvements in these more subjective measures did not clearly correlate to changes in hemoglobin (hematocrit) levels or absolute hemoglobin (hematocrit) values. Nor did they correlate with objective measurements of physical function or intermediate cardiac endpoints such a left ventricular function or anatomy. Finally none of the studies demonstrated durability of effect. For example, although the open-label CREATE study reported statistically significant higher scores in the higher target (and not necessarily achieved) hemoglobin group at one year, the difference disappeared by the following year.
Table 13: Quality of Life (QoL) Studies
iv. Emerging Signals of Harm
Several studies suggested that there might be unappreciated harm associated with ESAs.
Data from early surveys of the United States Renal Data System (USRDS)(1993-1999) were interpreted to mean that a higher hemoglobin level contributed to decreased mortality in dialysis patients.(Table 2)(Collins 1997, 2000, 2001, 2002, Ma 1999) Several societies, e.g., Canadian Society of Nephrology 1999, European Best Practice 2004, KDOQI 2007, UK Renal Association 2006, adopted treatment goals to achieve hemoglobin goals of 10 to 12 g/dl or greater. These USRDS data, however, did not reflect the natural history of the disease. Hematocrit (hemoglobin) data are typically entered into the system only in conjunction with Medicare claims for ESAs. (Koller direct review of USRDS files, Messana 2009) Many of the patients had been exposed to variable doses of erythropoietin, but the impact of this intervention was not addressed. In addition, the relatively small size of the cohorts with higher hematocrit levels and the limitations in extrapolating such data were not addressed.
Madore et al. conducted an analysis using census data from 21,899 patients at National Medical Care dialysis centers on January 1, 1993 and laboratory data for the antecedent three months. (Madore 1997) Complete laboratory data were available for 14,896. Descriptive statistics for parameters of interest were performed. The odds ratio for death increased progressively for hemoglobin levels below 10 g/dl. The odds risk associated with a hemoglobin of ≤ 8 g/dl was twice that associated with a hemoglobin between 10 and 11 g/dl. There was no survival benefit from achieved hemoglobin levels greater than 11 g/dl. Hemoglobin levels were inversely related to erythropoietin doses.
The Cotter group retrospectively analyzed the United States Renal Data System (USRDS) administrative claims data from 2000-2001 for 94,569 prevalent hemodialysis patients.(Cotter 2004, Zhang 2004) Patients were divided into cohorts on the basis of reported ESA dose and hematocrit(hemoglobin) at t = 0. Mortality over the next 12 months was assessed for each patient. Mortality was highest in those with the highest erythropoietin dose and the most severe anemia at baseline (Table 14).
Table 14: One Year Unadjusted Mortality (per 1,000 USRDS patients) by Hematocrit and Erythropoietin Dose Cohort (Zhang 2004)
Regidor et al assessed data from July 2001 to June 2003 for 58,058 patients dialyzed at the DaVita chain. (Regidor 2006) Information on co-morbid conditions was limited to that which could be extracted from the CMS Medical Evidence Form 2728. The results revealed increased mortality for patients with both higher and especially lower hemoglobin levels (Table 13). Trends were similar for unadjusted hazard ratios and ratios adjusted for case-mix differences and for incident and prevalent patients. Decline in hemoglobin levels over time was associated with increased mortality. The results also revealed disproportionately more mortality, both all cause and cardiovascular, for patients using higher doses of erythropoietin (Table 15). Baseline hemoglobin doses were higher in patients receiving the highest erythropoietin doses (Table 16).
Table 15: Case-Mix Adjusted Mortality Hazard Ratio by Hemoglobin Level (Regidor 2006)
Table 16: Mortality and Erythropoietin Dose (Regidor 2006)
Building on the Regidor and Cotter-Zhang analyses, Messana et al. retrospectively analyzed CMS Medical Evidence Form 2728 and Medicare claims data from 2002 to 2004 for 393,967 hemodialysis patients in a cross-sectional study. (Messana 2009) Mean quarterly hematocrit (hemoglobin) levels and erythropoietin/darbepoetin doses were determined (N = 2,712,197 patient-facility quarters). Case-mix adjustment was performed. 100,086 deaths were identified. Although they identified increased mortality at both high and low hematocrit levels, they observed a J-shape curve for mortality risk when dose was incorporated (Table 17). For any given hematocrit (hemoglobin) level, greater mortality was found with higher erythropoietin dosing. Co-morbidities were found to be an important factor in morbidity at low achieved hematocrit (hemoglobin) levels.
Table 17: Mortality Hazard Ratio (based on quarterly USRDS data) (Messana 2009)
Selinger et al. used the Veterans Affairs system data base to retrospectively assess the role of ESAs in acute stroke (CVA) in patients with estimated GFRs < 60 cm3/min per 1.73 m2 and hemoglobin levels < 12 g/dl using a case control design. (Selinger 2011) After adjustment for confounding variables, the likelihood of stroke was found to be greater in CKD patients using ESAs (odds ratio: 1.3) and even greater in CKD patients with cancer who used ESAs (odds ratio: 1.85). The median ESA dose was four time higher in CKD with cancer patients versus CKD patients without cancer whereas pre-treatment hemoglobin level did not differ.
v. Studies with Limitations
a—Scandinavian study by Furuland is sometimes cited as proof that the normalization of hemoglobin is safe. (Furland 2003, 2005a,b). This open-label study recruited a variety of renal patients (pre-dialytic, on peritoneal dialysis, and on hemodialysis) with mild anemia (hemoglobin levels between 9 and 12 g/dl without an exogenous ESA). It initially excluded patients with uncontrolled hypertension, diabetes, renal management problems, infection, inflammation, and cancer. Mid-study, after the results of the NCHT Besarab study were released, additional cardiac restrictions were added. 416 subjects were randomized into a 48 week (Finland, Iceland, Norway n = 163) or 76 (Sweden n = 253) week study in which entrants were dosed with erythropoietin to achieve a normal hemoglobin (13.5 -15 g/dl for women, 14.5-16 g/dl for men) or a subnormal level (9-12 g/dl). The death rate was reported to be equivalent for the normal hemoglobin and subnormal hemoglobin level treatment arms (Table 18). The study, however, was powered for exercise and not mortality.
Further evaluation of the cumulative mortality curves suggests that the mortality within each treatment arm was greater and occurred earlier for those who achieved lower hemoglobin levels. (Figure 9, Panels A and B) In addition, the drop-out rate was greater in the normal hemoglobin arm 56% versus the subnormal hemoglobin arms (43%) and greater at all time points resulting in a 5 week difference in study participation. The reasons for withdrawal differed for transplantation 14.8% versus 12% and adverse event/investigator decision 15.7% versus or 7.5% in the normal and subnormal hemoglobin treatment arms respectively. Although there were significant differences in erythropoietin doses by renal disease category and treatment arm cohort, there were no analyses assessing the role of erythropoietin dose in mortality and other causes for study withdrawal.
Table 18: Scandinavian Study
Figure 9: Scandinavian Study: Mortality Curves by Achieved Hemoglobin by Treatment Cohort
Panel A Normalized Hemoglobin
Panel B Subnormal Hemoglobin
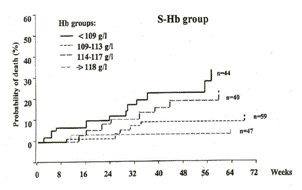
b—Other Studies Not Structured to Assess Long-term Safety
Many studies subsequent to the initial pivotal studies for approval of erythropoietin were not designed to assess long-term-safety and mortality. Many of these utilized active controls when comparing different routes of administration (subcutaneous or intravenous injection.) (Table 19) Many compared different ESAs (different active ingredient, different excipient, or different production-packaging technique) in either head-to-head or in switch studies (Table 20). Many utilized active controls when comparing different treatment regimens, e.g., hemoglobin targets or dosing frequency. (Table 21) Still others assessed the role of other concomitant treatments, e.g., EMLA cream, on the impact of ESA tolerability (Table 22). Many of the studies were relatively short in duration, six months or less. (Bahlmann 1991; n =129) Many of the studies were open-label. Many of the studies included less anemic populations. Few of the studies employed fixed dosing. None stratified by entry hemoglobin. Hemoglobin change, dose requirement, pain level, and patient satisfaction were frequent endpoints. Many of the studies, including several studies performed for regulatory approval, were equivalency or non-inferiority studies and presumed that studies of and (surrogate) endpoints for the predicate were adequate, that risk was equivalent for different patient populations, and that any safety issues were class-related (Tables 6, 7, and 8). Furthermore, the selection bias introduced by long screening periods and the inclusion of patients who were “washed-out” of from use another ESA (and not truly ESA-naïve) does not permit true assessment of drug response and adverse event incidence. Several of these studies remain unpublished (Table 3, Pivotal-Registration Studies section).
Table 19: Randomized Active Control Studies: Route of Administration
Table 20: Randomized Active Control Studies: ESA Type
Table 21: Randomized Active Control Studies: Different Treatment Regimens
Table 22: Randomized Active Control Studies: Other Study Types
Although these studies were not structured to assess long-term safety, safety signals emerged in at least two of the studies. In the open-label non-inferiority non-U.S.-based study comparing darbepoetin with the predicate in a 2:1 randomization over 32 weeks with a 20 week extension in 522 dialysis patients, a higher proportion of the 52 deaths that occurred (during the study or within the 28 day window of last drug dose or last assessment) were in the darbebpoetin arm 41/346 (12%) versus the erythropoietin arm 11/173 (6%); p = 0.06.(Tables 6,18) (Vanrenterghem 2002) Safety parameters seldom reach that level of statistical significance because studies are powered for efficacy, not safety. In addition, the descriptive data suggest that deaths occurred earlier in the darbepoetin arm, but that there was a convergence in cumulative mortality after an extended observation period (mean: two years). No survival curve was presented. Although these results were not replicated in the other pivotal study based by Nissenson et al, that U.S.-based study did differ by dosing.(Tables 6, 18) (Nissenson 2002) Darbepoietin and erythropoietin doses were twice that used in the Vanrenterghem study.
In open-label, non-inferiority studies comparing the new mimetibody, hematide, and the erythropoietin analogue, darbepoetin, in 983 pre-dialysis patients over 36 weeks with a 16-68 week extension period, there were differences in the cardiovascular composite safety endpoint that did not favor the study drug: 21.6% versus 17.1% (Table 8: PEARL study results). There were consistent difference in death (8.8% versus 6.7%), arrhythmia 5.6% versus 4%), and unstable angina (2.4% versus 0.9%). The cardiovascular adverse event disparity was greatest in the PEARL 2 study. Not all of the differences could be accounted for by imbalance at baseline. These safety risks were not balanced by reduction in transfusion risk. In the PEARL 2 study (n = 493), transfusions were required in more patients on hematide (11.4% in the initially low dose arm; 10.4% initially high dose) than on darbepoetin (4.8%). There were similar findings in the PEARL 1 study (n = 490) although they did not reach statistical significance.
Although it has been presumed that efficacy and adverse events associated with ESAs represent a class effect, we have been unable to find studies analyzing parameters that could assess and define risk differences between different ESAs.
vi. Studies to Assess Survival and/or Cardiovascular Endpoints
We have identified four studies that were structured to assess survival and/or cardiovascular endpoints: the Correction of Hemogloblin and Outcomes in Renal Insufficiency (CHOIR) trial, Cardiovascular Risk Reduction by Early Anemia Treatment with Epoetin Beta (CREATE), the Normalization of Hematocrit Trial (NCHT), and the Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) (Table 23). All were designed to assess target hemoglobin levels-although the targets differed by study. None were designed to assess dose effect in any of the three ESAs evaluated (erythropoietin-alpha: CHOIR and NCHT; erythropoietin-beta: CREATE; darbepoetin: TREAT). All were designed to follow patients for at least one year. All recruited more than 500 patients. Three of the studies were open-label (CHOIR, CREATE, and NCHT). Three of these studies were conducted in pre-dialysis patients (CHOIR, CREATE, and TREAT). Renal status for inclusion in these three studies was determined using glomerular filtration rates (GFR), but these rates were not measured directly or with the use of concomitant serum and urinary creatinine values. They were estimated using serum creatinine values in formulas (Cockcroft– Gault for CREATE [inclusion range 15.0-35.0 ml/min/1.73 m2]; Modification of Diet in Renal Disease for CHOIR [inclusion range 15-50 ml/minute/1.73 m2] and TREAT [inclusion range 20-60 ml/minute/1.73 m2 ]. Patients with anemia not attributable to renal disease were included. (See Anemia Background section.) Two studies specifically recruited patients with either cardiac disease (NCHT) or type 2 diabetes with its known likelihood of macrovacular disease (TREAT).
No studies stratified patients on the basis of ESA-naïve hematocrit (hemoglobin) levels, ESA doses or dialysis adequacy/renal clearance. One study (TREAT) stratified patients using urinary protein:creatinine ratios because of its putative value for cardiovascular disease. (Hemmelgarn 2010, Keane 2003) No study included criteria for red blood cell transfusion. No study collected data on the reason for red blood cell transfusion (anemia management versus other indication e.g. surgical procedure or GI bleed), the pre-transfusion hematocrit (hemoglobin) level, or the number of units transfused. The reported data are limited to numbers of patients transfused. Drop-out rates were high (CREATE 21% overall, 25% high target, 17% low target; CHOIR 46% overall, high target: death 7%, other 21%, non-fatal primary event 10%, renal replacement therapy 18%, low target death 5%, other 22%, non-fatal primary event 8% renal replacement therapy 16%; TREAT high target: death 20%, death in 30 day window after closure 0.4%, non-fatal primary event 2%, other 11%, treatment stopped-continued in study 21%, low target: death 20%, death in 30 day window after closure 0.5%, non-fatal primary event 2%, other 11%, treatment stopped-continued in study 22%). Drop-out rates were not reported for the NCHT. Two of the studies were terminated early (NCHT and CHOIR). The NCHT was halted by the safety monitoring board because a divergence in survival, not in favor of the higher hemoglobin treatment arm. The trial was stopped before statistical significance could be reached. The CHOIR trial was halted by the monitoring board because it was thought to be unlikely that benefit for the high hemoglobin target would be demonstrated. The trial was stopped before statistical significance for futility could be reached.
The studies were able to achieve hemoglobin (hematocrit) separation between the high and low target arms in all studies. Not all patients within the target arms achieved the desired targets despite the individualized titration. Indeed, in CHOIR, the doses for those patients who achieved the target hemoglobin (whether high or low) were lower than those who failed to achieve the target hemoglobin: 10.5-11 g/dl target: total cohort mean 6276 U/wk, achieved 6057 U/wk, not achieved 11,098 versus 13-13.5 g/dl target: total cohort mean 11,215 U/wk, achieved 10694 U/wk, not achieved 12,884 U/wk. (See Table 23 for original and amended target values.) (This heterogeneity in response in non-fixed dose studies has confounded attempts to determine whether hemoglobin levels or drug exposure parameters, e.g., peak dosage, cumulative dose, or peak serum dose via various dose regimens and/or routes of administration, are contributing factors to adverse events and whether different mechanisms underlie different adverse events.)
None of the trials demonstrated any benefit for either survival or decreased morbidity from cardiovascular events (Tables 23 and 28) Indeed, review of the actual numbers of events suggests that there was a trend to more deaths and cardiovascular events in the higher target arms—although stroke (cardiovascular accident = CVA) accounted for most of these events in the TREAT trial. There was also a trend to more cancer deaths in the higher target treatment arm of TREAT. This occurrence in patients with a history of cancer, but no known active oncologic disease at the time of enrollment, suggests a tumor “promoter” role and is consistent with that which was seen in oncology patients using ESAs on a more intermittent basis. (See NCD CAG-0383N.) Although these results were unexpected, they are consistent and provide a strong safety signal. Statistical significance would not be expected because the studies were designed and powered for a different hypothesis and two were terminated early. The absence of definitive proof of harm cannot be used to establish absence of risk. (See NCD CAG-0383N for studies with nascent negative outcomes that were terminated or remained unpublished.)
Table 23A: Studies Designed to Assess Survival and/or Cardiovascular Outcomes
Table 23B: Studies Designed to Assess Survival and/or Cardiovascular Outcomes (continued)
Table 23C: Studies Designed to Assess Survival and/or Cardiovascular Outcomes (continued)
c. Systematic Reviews
We are aware of several published systematic reviews of erythropoiesis stimulating agents, anemia, and/or transfusions and describe them below briefly. Systematic reviews are based on a comprehensive search of published materials to answer a clearly defined and specific set of clinical questions. A well-defined strategy or protocol (established before the results of individual studies are known) is optimal.
i. Cochrane Collaboration
aa. Transfusion thresholds and other strategies for guiding allogeneic red blood cell transfusion (Review)
Carless PA, Henry DA, Carson JL, Hebert PPC, McClelland B, Ker K
Publication status and date: Edited (no change to conclusions), published in Issue 10, 2010. Review content assessed as up-to-date: 31 July 2009.
“…Most clinical practice guidelines recommend restrictive red cell transfusion practices, with the goal of minimising exposure to allogeneic blood (from an unrelated donor). The purpose of this review is to compare clinical outcomes in patients randomised to restrictive versus liberal transfusion thresholds (triggers)…Restrictive transfusion strategies did not appear to impact on the rate of adverse events compared to liberal transfusion strategies (i.e. mortality, cardiac events, myocardial infarction, stroke, pneumonia and thromboembolism). Restrictive transfusion strategies were associated with a statistically significant reduction in the rates of infection (RR=0.76; 95% CI 0.60 to 0.97). The use of restrictive transfusion strategies did not reduce hospital or intensive care length of stay…The existing evidence supports the use of restrictive transfusion triggers in patients who are free of serious cardiac disease. The effects of conservative transfusion triggers on functional status, morbidity and mortality, particularly in patients with cardiac disease, need to be tested in further large clinical trials. For most patients, blood transfusion is probably not essential until haemoglobin levels drop below 7.0 grammes per decilitre…”
Comment: The review has not been updated to include major studies including TRACS (Hajjar 2010; n = 512) and the most recent FOCUS data (Carson 2009-abstract; 2011).
bb. Haemoglobin and haematocrit targets for the anaemia of chronic kidney disease (Review)
Strippoli GFM, Navaneethan SD, Craig JC, Palmer SC
Publication status and date: Edited (no change to conclusions), published in Issue 2, 2010. Review content assessed as up-to-date: 14 August 2006.
“…Full-text assessment of 49 potentially eligible papers identified 22 eligible trials (3707 patients) (Abraham 1990; Bahlmann 1991; Berns 1999; Besarab 1998; Brandt 1999; Canadian 1991;Clyne 1992; Conlon 2000; Foley 2000; Gouva 2004; Kleinman 1989; Kuriyama 1997;Levin 2005; Lim 1989;Morris 1992; Parfrey 2005; Revicki 1995; Roger 2004a; Scandinavian 2003; Sikole 1993; Teehan 1991; Watson 1990)…Twenty two trials (3707 patients) were included. In general study quality was poor. There is a need for more adequately powered, well-designed and reported trials. Trials should be pragmatic, focusing on hard endpoints (mortality, ESKD, major side effects) or outcomes which were previously not studied adequately (e.g. seizures, quality of life). In general study quality was poor. There is a need for more adequately powered, well-designed and reported trials. Trials should be pragmatic, focusing on hard endpoints (mortality, ESKD, major side effects) or outcomes which were previously not studied adequately (e.g. seizures, quality of life).”
Comment: The review has not been updated to include major studies including CREATE (Drueke 2007; n = 603), CHOIR (Singh 200; n = 1432), and TREAT (Pfeffer 2009; n = 4038)
cc. Recombinant human erythropoietin for chronic renal failure anaemia in pre-dialysis patients (Review)
Cody JD, Daly C, Campbell MK, Khan I, Rabindranath KS, Vale L, Wallace SA, MacLeod AM, Grant A, Pennington S
Publication status and date: Edited (no change to conclusions), published in Issue 1, 2009. Review content assessed as up-to-date: 24 May 2005.
“…The review now includes 15 trials (Abraham1990; Brown 1995; Clyne 1992; Eschbach 1989; Ganguli 2003; Kleinman 1989; Kuriyama 1997; Lim 1989; Roth 1994; Stone 1988; Teehan 1989; Teehan 1991; Teplan 2001; Teplan 2003; Watson 1989) with a total of 461 participants. Twelve trials were reported in full published papers, and three reported in abstract form only (Brown 1995; Ganguli 2003; Teplan 2001). The degree of renal function was broadly similar amongst the participants of the trials with the exception of Teplan 2003 where renal failure was less advanced. It was subsequently confirmed that four of these studies (Abraham 1990; Eschbach 1989; Lim 1989; Stone 1988) formed part of a larger multicentre trial (Teehan 1991)...Fifteen trials (461 participants) were included. Treatment with rHu EPO in pre-dialysis patients corrects anaemia, avoids the requirement for blood transfusions and also improves quality of life and exercise capacity. We were unable to assess the effects of rHu EPO on progression of renal disease, delay in the onset of dialysis or adverse events. Based on the current evidence, decisions on the putative benefits in terms of quality of life are worth the extra costs of pre-dialysis rHu EPO need careful evaluation…” The excluded studies are listed on page 38.
Comment: The review does not include major studies including CREATE (Drueke 2007; n = 603), CHOIR (Singh 200; n = 1432), and TREAT (Pfeffer 2009; n = 4038).
ii.National Institute for Clinical Excellence (NICE)
NICE does not conduct assessments/reviews of transfusions because “this procedure does not fall within the Institute's definition of an interventional procedure. To fall within the Programme's remit, a notified procedure must involve an incision or a puncture or entry into a body cavity, or the use of ionising, electromagnetic or acoustic energy.”
iii.Serious Hazards of Transfusions (SHOT)
An independent haemovigilance group funded by the UK Blood Services (NHS Blood and Transfusion, Northern Ireland Blood Transfusion Service, Scottish National Blood Transfusion Service, Welsh Blood) and affiliated with the Royal College of Pathologists.
Annual Review 2009 (www.shotuk.org/wp-content/uploads/2010/06/SHOT-2009-Summary.pdf; www.shotuk.org/wp-content/uploads/2010/07/SHOT2009.pdf; accessed 11/28/2010.)
Cohen H, Mold D, Jones H, Davies T, Mistry H, Ball J, Asher D, Cawley C, Chaffe B, Chapman C, Gray, Jones J, Milkins C, New H, Norfolk D, Regan F, Still E, Tinegate H, Taylor C.
Deaths from transfusion have declined over time to less than 10% of those in 1996-1997 (Table 25). Red blood cell transfusions decreased to 80% of those in 1999-2000 (Table 24).
Table 24: Secular Trends in Blood Usage in the United Kingdom
Table 25: Adverse Events with Blood Usage in the United Kingdom
4. MEDCAC
A Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) meeting was convened on this issue on March 24, 2010. Chronic renal disease and anemia management with erythropoietic stimulating agents were reviewed discussed. (www.cms.gov/medicare-coverage-database/details/medcac-meeting-details.aspx?MEDCACId=52&bc=BAAQAAAAAAAA&; accessed July 19, 2010.)
A second Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) meeting was convened on this issue on January 19, 2011. At the request of the panelists on the March 2010 MEDCAC, renal transplantation and the impact of red blood cell transfusion were reviewed. (www.cms.gov/medicare-coverage-database/details/medcac-meeting-details.aspx?MEDCACId=57&bc=BAAQAAAAAAAA&; accessed January 21, 2011.)
5. Evidence-based guidelines
a. American Medical Directors Association in conjunction with representatives from the American Association of Homes and Services for the Aging, the American College of Health Care Administrators, the American Geriatrics Society, American Health Care Association, the American Society of Consultant Pharmacists, National Association of Directors of Nursing Administration in Long-Term Care, National Association of Geriatric Nursing Assistants, and the National Conference of Gerontological Nurse Practitioners
Anemia in the Long-term Care Setting 2007. NGC:005655 Guidelines not on website Hardcopy on file with CMS.
“…The World Health organization defines anemia as a hemoglobin of less than 12 g/dl in women and less than 13 g/dl in men…Anemia is a marker for increased morbidity, hospitalizations, mortality, and health care costs…Caregivers and health care professionals may not relate non-specific symptoms such as fatigue, weakness, and lack of stamina to anemia…Anemia associated with chronic kidney disease (CKD) was redefined in 2006 by the National Kidney Association (NKF) Kidney Disease Outcomes Quality Initiative (K/DOQI) as hemoglobin of less than 12 g/dl in women and less than 13.5 g/dl in men in the presence of renal dysfunction...Anemia associated with CKD can be severe and may lead to cardiovascular complications and death...Deficiency of the hormone erythropoietin is the primary but may not be the sole cause of anemia associated with CKD…Synthetic erythropoietin-stimulating agents (ESAs) are available to treat this type of anemia…The use of ESAs to treat anemia associated with CKD should be carefully evaluated in frail elderly patients in the longterm care setting…”
The authors state that they were an interdisciplinary group. The guideline provides an algorithm for assessment, treatment, and monitoring anemia although the authors did not do a comprehensive review of primary data, grade evidence, or provide a rating scheme for the strength of the recommendations. For example, the WHO criteria were developed for epidemiologic surveillance of nutrient, especially iron, deficiencies in third world settings. For example, anemia is not erythropoietin-mediated until after the initiation of dialysis (Radtke 1979). For example, the text implies that ESAs will improve hematocrit level, transfusion need, quality of life, exercise performance, and cognitive function based on only two cited studies with 11 and 23 patients respectively (Bedani 2001 and Moreno 1996) which were not critically assessed. The report of an 8 g/dl improvement in older patients does not appear to match the values in figure 1 (Moreno 1996 [reference 57]) and does not address the impact of drop-out (14% for total ESA treated population). The guideline can only be obtained at a cost of $15. Funding for the guideline was supported by the following: Amgen, Merck & Co., Inc., Ross Products Division of Abbott Laboratories, and Sanofi-Aventis.
b. British Committee for Standards in Haematology
Guidelines for the Clinical Use of Red Cell Transfusions 2001 (British Journal of Haematology 113: 24-31) Currently under revision
Blood Transfusion Task Force (Kelsey P, Boulton F, Bruce M, Cohen H, Duguid J, Knowles SM, Murphy MF, Poole G, Williamson LM, Wallington TB. Reviewed by the Royal College of Surgeons of England, the Royal College of Physicians, and the Royal College of Anaesthetists.
“…Red cell transfusion when estimates of actual and anticipated haemoglobin concentrations are >10 g/dl. Red cell transfusion is indicated when the haemeglobin concentration is < 7 g/dl. Red cell transfusions should be given in relation to the rate of ongoing red cell loss…The correct strategy for transfusion of patients with haemaglobin concentrations between 7 and 10 g/dl is less clear. Clinicians often transfuse red cells, although available evidence suggests that this is often not justified. In patients who may tolerate anaemia poorly, e.g., patients over the age of 65 years and patients with cardiovascular or respiratory, consider adopting a higher concentration at which transfusions are indicated, e.g., when the haemoglobin concentration becomes < 8 g/dl. … In principle, red cell transfusions for patients with chronic aenemia should be given at intervals to maintain the haemoglobin just above the lowest levels associated with symptoms of anaemia, but it may be difficult to determine what this is for individual patients…”
Guideline on the Administration of Blood Components 2009. (www.bcshguidelines.com; accessed12/01/2010)
Harris A, Atterbury C, Chaffe B, Elliott C, Hawkins T, Hennem S, Howell C, Jones J, Murray S, New H, Norfolk D, Pirie L, Russell J, Taylor C.
The purpose of this guideline is to provide national guidance on pre-transfusion blood sampling and the prescription, requesting, collection and administration of blood components to adults, children and neonates in order to provide a basis for the development of standardised local guidelines and practice.
c. Canadian Society of Nephrology
Guidelines for the management of chronic kidney disease 2008. (Canadian Journal of Medicine)
Levin A, Hemmelgarn B, Culleton B, Tobe S, McFarlane P, Ruzicka M, Burns K, Manns B, White C, Madore F, Moist L, Klarenbach S, Barrett B, Fole Ry, Jindal K, Senior P, Pannu N, Shurraw S, Akbari A, Cohn A, Reslerova M, Deved V, Mendelssohn D, Nesrallah G, Kappel J, Tonelli M.
“…Anemia is prevalent among patients with an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Anemia is associated with adverse outcomes in patients with chronic kidney disease, including hospital admission, cardiovascular disease and mortality. Although erythropoietin deficiency is a well-known cause of anemia in this population, the guidelines recommend that other potential causes of anemia should be sought (e.g., iron deficiency) and treated accordingly. To date, therapies to normalize the hemoglobin level in these patients have not shown any health benefit. These therapies have been associated with an increased incidence of death or need for dialysis. Based on this evidence, a target hemoglobin level of 110 g/L is recommended for patients with chronic kidney disease (acceptable range 100 – 120 g/L). The use of erythropoiesis-stimulating agents for the treatment of anemia in patients with chronic kidney disease is associated with potential adverse outcomes, including increased blood pressure and thrombotic complications. They should be prescribed by a specialist with experience in prescribing these agents. Iron therapy is an important component of anemia management. We recommend that the oral form of iron be considered preferentially over the intravenous form…”
d. Caring for Australasians with Renal Impairment-Australian and New Zealand Society of Nephrology
Erythropoietin 2004 (www. cari.org.au/CKD_Prevent_List_Published/Erythropoietin.pdf accessed12/01/2010)
Johnson D.
The weight of clinical evidence indicates that erythropoietin exerts neither a beneficial nor deleterious effect on the progression of renal impairment in patients with chronic renal insufficiency. (Level II Evidence, 6 small randomised controlled trials; clinically relevant outcomes; inconsistent effects) Of the 6 RCTs published to date, 5 trials have found no significant effect of erythropoietin administration on the progression of CKD. One trial with significant flaws observed that erythropoietin significantly retarded renal failure progression, primarily in non-diabetics.
Biochemical and haematological targets. Haemoglobin. 2008 (Nephrology)
McMahon L.
“The targeting of haemoglobin concentrations above 13 g/L has been associated with an increased mortality in chronic kidney disease (CKD) patients (dialysis and pre-dialysis) and is therefore currently considered inadvisable (Level I evidence)”.
“…Substantial evidence now indicates that targeting (without necessarily achieving) haemoglobin concentrations above 130 g/L with ESA in patients with CKD results in an increased mortality and morbidity with little benefit (compared to targeting concentrations below 120 g/L) and at a higher cost. Older patients and those with more advanced cardiovascular disease and/or diabetes are at highest risk, which appears to pertain to both dialysis and pre-dialysis patients. In addition, there appears to be an increased risk of hypertension and arteriovenous access thrombosis in patients targeted for higher haemoglobin concentrations without substantial evidence of benefit in quality of life or normalisation of exercise capacity. The ESA dosage and associated cost of achieving and maintaining higher haemoglobin concentrations is significantly greater…”
The other clinical recommendations were based primarily on Level 3 (case control or cohort) or level 4 (cases series) evidence.
e. College of American Pathologists (CAP)
This professional group no longer issues transfusion practice guidelines although they have done so in the past.
f. European Blood Alliance
Manual of Optimal Blood Use: Support for Safe, Clinically Effective Use of Blood in Europe 2010 (www.optimalblooduse.eu; accessed12/15/2010)
McClelland DBL, Pirie E, Franklin IM for the EU Optimal Blood Use Project Partners;
Co-funded by the European Commission and the Scottish National Blood Transfusion Service
Published by the Scottish National Blood Transfusion Service
“…Optimal use of blood is defined in this manual as “The safe, clinically effective and efficient use of donated human blood.” However, for many of the familiar and widely accepted indications it is a fact that there is surprisingly little high quality evidence to establish the effectiveness of transfusion therapy. As a result, clinical transfusion guidelines must often be based on inadequate information. Information in this chapter about the quality and grading of evidence for clinical practice guidelines has been drawn from the German Guidelines for Therapy with Blood Components and Plasma Derivatives (2009.) Another useful sources (sic) is the database of systemic reviews at the website www.transfusionguidelines.org.uk. Studies in several European countries show that although patients undergoing surgery and treatment for malignant disease are major users of transfusion, a substantial portion of all transfusions are used for patients who do not belong to any simple category, who are in older age groups and who have essentially “medical” conditions, often with multiple diagnoses, interventions, and episodes of hospital care. …Decision-making can be relatively straightforward when a patient has a life-threatening major haemorrhage, bleeding associated with profound thrombocytopenia, or severe, disabling symptoms of anaemia associated with cancer chemotherapy. The decision is much less clear – for example in an elderly patient, who has a haemoglobin concentration of 80g/l, has no evident symptoms of anaemia, is haemodynamically stable and is not bleeding….”
The following information in the Alliance manual is based on the German Medical Association’s cross sectional guidelines for therapy with blood components and plasma derivatives in Bundesaertztekammer 2009, 4th revised edition (Table 26).(Heim 2009) The information presumes that the patient is not hemoconcentrated and not hypovolemic.
Table 26: Transfusion Guidance and Evidence Rating
g. European Renal Association-European Dialysis and Renal Transplant Association.
European RenalBestPractice
Anaemia management in patients with chronic kidney disease: a position statement by the Anaemia Working Group of European RenalBestPractice (ERBP) 2009
Locatelli F, Covic B, Eckardt K-U, Wiecek A, Vanholder R, ERA-EDTA ERBP Advisory Board: Abramovicz D, Cannata Andia J, Cochat P, Fouque D, Heimburger O, Jenkins S, Lindley E, London G, MacLeod A, Marti A, Spasovski G, Tattersall J, Van Biesen W, Wanner C, Zoccali C.
“…A specially appointed ERA-EDTA Work Group met in Paris to discuss European guideline planning in early January 2008, and agreed that the Association should continue producing and updating guidelines in collaboration with KDIGO. It also agreed that ERA-EDTA should issue suggestions for clinical practice in areas in which evidence is lacking or weak, which will be presented as ‘position statements’ rather than clinical guidelines. It was also decided to issue position statements about guidelines (recommendations issued by other bodies, of which the current publication is the first result). Finally, the group opted to change the name EBPG to European Renal Best Practice (ERBP) as a means of acknowledging that, especially in nephrology, it is difficult to generate real ‘guidelines’ because of the lack of sufficient evidence. In this context, and while awaiting the publication of the KDIGO anaemia guidelines possibly in 2011, an ad hoc work group was commissioned by the ERBP Advisory Board to give its opinion on the ‘hot topic’ of Hb targets, including recently raised issues that were not covered by KDOQI in 2006…”
Regarding the definition of anemia, “...In 2006, KDOQI modified this definition by giving a single criterion for diagnosing anaemia in adult males (Hb < 13.5 g/dl, regardless of age) because the decrease in Hb among males aged > 60 years is often attributable to concurrent diseases. The ERBP Work Group agrees with this new definition…In the opinion of the ERBP Work Group, it appears reasonable to maintain the lower limit of the target, although the actual evidence for choosing this value is also very limited. On the basis of new evidence, Hb values of 11 – 12 g/dl should be generally sought in the CKD population without intentionally exceeding 13 g/dl…The ERBP Work Group believes that there is a need for better understanding as to whether any harm may be associated with attempts to reach higher Hb values in patients with comorbidities or those who are hyporesponsive to ESAs…The ERBP Work Group agrees with the recent position of KDIGO that the available quality of life data vary in quality and are often inconclusive. In the opinion of the ERBP Work Group, ESA therapy should be cautiously used in patients with CKD and malignancies as no information is available concerning the risk of mortality and tumour growth in this subset of patients...”
“…In the opinion of the group, epoetin delta should be administered similarly to epoetin alpha…The ERBP Work Group considers the safety and tolerability of CERA to be similar to that of other ESAs…The ERBP Work Group recommends stringent pharmacovigilance for biosimilars of epoetin alpha that can be administered only intravenously…”
The workgroup did not consider the TREAT study which they indicated was ongoing at the time of the guideline discussions.
Target haemoglobin to aim for with erythropoiesis-stimulating agents: a position statement by ERBP following publication of the Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Study.
Locatelli F, Aljama P, Canaud B, Covic A, De Francisco A, MacDougall IC, Wiecek A, Vanholder R, and on behalf of the Anaemia Working Group of European Renal Best Practice (ERBP)
"…The aim of this second position statement on anaemia is to give guidance on the interpretation of the recently published Trial to Reduce Cardiovascular Events with Aranesp® Therapy (TREAT) Study [Pfeffer 2009], and its possible relevance to recommended treatments and Hb targets to be used when treating chronic kidney disease (CKD) patients with erythropoiesis-stimulating agents (ESA) therapy, while awaiting the publication of KDIGO anaemia guidelines (probably sometime in the first half of 2011).
This paper is not intended to represent a new guideline as it is not the result of a systematic review of the evidence…."
"…The TREAT Study was conducted in CKD patients with type 2 diabetes, and the debate now is whether the data can only inform anaemia management in this specific patient population. Some scientific purists might argue that evidence from the TREAT Study is applicable only to patients with type 2 diabetes and CKD stages III to IV. Others will argue that, although the TREAT Study was conducted in type 2 diabetics, the results on strokes and malignancies (although secondary analyses) are concordant with a large body of other evidence, such as that obtained from the US Normal Hematocrit Trial [Kilpatrick 2008] and the CHOIR Study [Szczech 2008], not to mention multiple oncology trials, all of which suggest possible concerns about increased thrombogenicity with ESA therapy when targeting higher Hb levels than suggested by present guidelines. At the very least, greater caution should be exerted in using high ESA doses, and targeting Hb levels >12 g/dL, in type 2 diabetics [Goldsmith 2010, Locatelli 2010].
Although there is no 'grade A' evidence to suggest that possible concerns about thrombotic adverse events are dose-related, post hoc analyses from both the US Normal Hematocrit Trial [Kilpatrick 2008] and the CHOIR Study [Szczech 2008] suggested that the patients who had the worse outcomes were those who were the most resistant to treatment, and who were receiving the highest doses of ESA. Thus, while this ERBP group still maintains a view that 'Hb values of 11-12 g/dL should be generally sought in the CKD population without intentionally exceeding 13 g/dL [Locatelli 2009; see above.], their opinion is that the doses of ESA therapy to achieve this level of target haemoglobin should also be considered. If a patient can obtain a Hb of 11.5 g/dL on no or low doses of ESA therapy, then there is less cause for concern than with a patient who is requiring very high doses of ESA therapy to achieve this Hb…."
h. Kidney Disease Improving Global Outcomes (KDIGO) (managed by the National Kidney Foundation)
KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for Anemia in Chronic Kidney Disease: 2007 Update of Hemoglobin Target (www.kidney.org/professionals/kdoqi/guidelines_anemiaUP/index.htm accessed 11/28/2010)
VanWyck D, Eckardt K-U, Adamson J, Berns J, Eckardt K-U, Fishbane S, Foley R, Ghaddar S, Gill J, Jabs K, Bargo McCarley P, Nissenson A, Obrador G, Stivelman J, White C. Liaison Members Locatelli F, Macdougall IC. Evidence Review Team National Kidney Foundation Center for Clinical Practice Guideline Development and Implementation at Tufts-New England Medical Center
(KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Anemia in Chronic Kidney Disease 2006) www.kidney.org/professionals/kdoqi/guidelines_anemia/guide1.htm; accessed 11/28/2010)
(Adamson J, Bailie G, Berns J, Fishbane S, Foley R, Ghaddar S, Gill J, Jabs K, Bargo McCarley P, Messner H, Nissenson A, Obrador G, Stivelman J, White C.
“…In the opinion of the Work Group, in dialysis and nondialysis patients with CKD receiving ESA therapy, the selected Hb target should generally be in the range of 11.0 to 12.0 g/dL…In dialysis and nondialysis patients with CKD receiving ESA therapy, the Hb target should not be greater than 13.0 g/dL…”)
Currently this clinical guideline is undergoing revision. “Anemia in CKD” is under the leadership of Drs. Patrick Parfrey and John McMurray (Anticipated Publication – 2012)
i. National Collaborating Centre for Chronic Conditions.
Anaemia management in chronic kidney disease. National clinical guideline for management in adults and children.
London (UK): Royal College of Physicians; 2006. 172 p. [295 references]
“…Patients with CKD should be evaluated for risk stratification of cardiovascular disease. Patients with CKD should be assessed for cardiovascular risk including fasting lipid profile, blood pressure, tobacco use (smoking) history, family history of premature cardiovascular disease, obesity, and physical activity level. Strategies to reduce cardiovascular risk factors should be implemented. Consider treatment of anemia in patients with CKD with an erythropoietic stimulating agent if the hemoglobin is less than < 10 g/dL and after appropriate evaluation and ruling out other possible causes. Such treatment may require referral to nephrology or hematology and more frequent monitoring of hemoglobin values...” Adverse events were listed: “…Hypertension occurs in 20 to 30 percent of patients and is easily treatable. Vascular access thrombosis. Hyperkalemia. Myalgia and flu-like symptoms. Injection pain and skin irritation around the injection site. Pure red cell aplasia is very rare and is associated with anti-erythropoietin antibodies…” The following evidence table is based on the evidence table in the guidelines (Table 27).
Table 27: NCCCC Anemia Management Guidelines for Patients with Renal Disease
j. National Institute for Health and Clinical Excellence (NICE)
Anaemia Management in People with Chronic Kidney Disease: Clinical Guideline: Rapid Update of Guideline #39 (Accessed July 21, 2010 and March 1, 2011)
“Consider investigating and managing anaemia in people with CKD if: their Hb levels falls to 11 g/dl or less (or 10.5 g/dl if younger than 2 years) or they develop symptoms attributable to anaemia (such as tiredness, shortness of breath, lethargy, and palpitations.” (No grade assigned) (p 41)
“ESAs need not be administered where the presence of comorbidities, or the prognosis, is likely to negate the benefits of correcting the anaemia.” (Grade D) (p42)
“Treatment with ESAs should be offered to people with anaemia of CKD who are likely to benefit in terms of quality of life and physical function.” (Grade A) (p43)
“A trial of anaemia correction should be initiated when there is uncertainty over whether the presence of comorbidities, or the prognosis, would negate the benefit from correcting the anaemia with ESAs.” (Grade D) (p42)
“The correction to normal levels of Hb with ESAs is usually not recommended in people with anaemia of CKD. Typically maintain the aspirational Hb range between 10 and 12 g/dl for adults, young people and children aged 2 years and older, and between 9.5 and 11.5 g/dl for children younger than 2 years of age, reflecting the lower normal range in that age group. To keep the Hb level within the aspirational range, do not wait until Hb levels are outside the aspirational range before adjusting treatment (for example, take action when Hb levels are within 0.5 g/dl of the ranges’s limits).” (No grade assigned) (p44)
“Where a trial of ESA therapy has been performed, the effectiveness of the trial should be assessed after an agreed interval. Where appropriate, a mutual decision should be agreed between the clinician, the person with anaemia of CKD and their families and carers on whether or not to continue ESA.” (Grade D) (p42)
“All people started on ESA therapy should be reviewed after an agreed interval in order to decide whether or not to continue using ESAs” (Grade D) (p42)
“After other causes of anaemia, such as intercurrent illness or chronic blood loss have been excluded, people with anaemia of CKD should be considered resistant to ESAs when: an aspirational Hb range is not achieved despite treatment with ≥ 300 IU/kg/week of subcutaneous epoietin or ≥450 IU/kg/week of intravenous epoietin or 1.5 ug/kg/week of darbepoetin, or there is a continued need for the administration of high doses to maintain the aspirational Hb range.” (Grade D) (p44)
Comments: Recommendations without the usual accompanying evidence grade assessments or low grade assessments raise questions about the validity of the recommendation. The basis for the 10-12 g/dl aspiration goal appears to be based on the Collins group retrospective assessments mortality and hemoglobin based on USRDS data (NICE reference 60). The USRDS data do not reflect the natural history of anemia because hematocrit (hemoglobin) levels infrequently enter the system unless they are on a billing claim for ESAs. The presence, magnitude, and impact of an intervention (ESA) on outcomes were not addressed in the papers.
k. World Health Organization (1994)
Indicators and Strategies for Iron Deficiency and Anemia Programmes. Report of the WHO/UNICEF/UNU Consultation. 1994
Achadi E, El Amin A, Florentino R, Galil A, Hallberg L, Suboticanes-Buzina K, Thwin A, Viteri F, Walter T, Wenzhen C, Harrison K, Kachondham Y, Zavaleta N, Clay W, Dirren H, Parr R, Robinett D, Seifman R, Simon S, Theuer R, Yip R, Alnwick D, Scrimshaw N, Antezana F, Bailey K, Benbouzid D, Buzina R, de Benoist, B, Herrman J, Johnson R, Savioli L, Underwood B, Van der Pols J, Verster, A. Conference in Geneva, Switzerland, 6–10 December, 1993.
The World Health Organization (WHO) definitions for anemia were developed for surveillance of anemia due to nutritional deficiency and parasitic infections. Anemia was defined to present at sea level with hemoglobin levels < 13 g/dl in adult men, < 12 g/dl in non-pregnant adult women, < 11 g/dl in pregnant adult women, < 12 g/dl in children aged 12-14 years < 11.5 g/dl in children 5-11 years, and < 11 g/dl in children 6 to 59 months. The report notes that “It is well known that normal haemoglobin distributions vary with age and gender, at different stages of pregnancy, and with altitude and smoking” (Chanarin 1971, Hurtado 1945). “There is also evidence of a genetic influence. In the United States, for example, individuals of African extraction have haemoglobin values 5 to 10 g/l lower than do those of European origin. This contrast is not related to iron deficiency” (Perry 1992)… “Annex 3 provides age-related criteria for normal haemoglobin and haematocrit levels developed by the Centers for Disease Control and Prevention in Atlanta, USA “(Expert Scientific Working Group. AJN 1985). “Criteria for stages of pregnancy, and adjustment factors for altitude and smoking are also provided. For populations of African extraction, recent analysis indicates that achieving a similar screening performance (sensitivity and specificity) requires a haemoglobin criterion that is 10 g/l (0.62 mmol/l) lower than those shown in Table 6” (Johnson-Spear 1994, Yip 1997)…”Severe anaemia in pregnancy is defined as haemoglobin <70 g/l and requires medical treatment. Very severe anaemia is defined as haemoglobin <40 g/l. Very severe anaemia in pregnant women is a medical emergency due to the risk of congestive heart failure; maternal death rates are greatly increased….”
6. Professional Society Position Statements
Various professional societies expressed positions via submitted public comment.
a. The American Society of Nephrology (ASN) believe that current ESAs may be dangerous if used for overly aggressive treatment targets compared with practices that are compatible with current treatment guidelines. They also believe that continued access to ESAs is required to give both dialysis and non-dialysis patients with CKD, a better chance at receiving and maintaining the function of a kidney transplant.
b. The National Kidney Foundation (NKF) believes that the anemia target should be generally consistent with the recommendation in the 2007 Kidney Disease Outcomes Quality Initiative (KDOQI) Update of Hemoglobin (HB) Target, and the FDA package inserts for the three approved ESAs, i.e. a range from 10 to 12 g/dL.
7. Expert Opinion
CMS sought and received expert opinion through the TA and MEDCAC processes.
8. Public Comments
Public comment sometimes cites the published clinical evidence and gives CMS useful information. The CMS uses the initial public comments to inform its proposed decision. The CMS responds in detail to the public comments on a proposed decision when issuing the final decision memorandum.
Initial Comment Period: 6/16/10 – 7/16/10
During this public comment period, the CMS received a total of nine timely comments. Of these public comments received, five were from professional organizations, two were from marketers of ESAs, one was from a professional society and one was from a patient advocacy group.
The majority of comments suggest that policies should align with FDA approved labeling and that policies should differentiate between dialysis patients and non-dialysis patients. Some commenters suggested that quality of life (QoL) should be considered an important patient outcome. Other commenters were concerned with the effect the new prospective payment system (PPS) may have on the treatment of anemia. However, the PPS is outside the scope of this NCD.
One commenter supported CMS’ efforts to review the available clinical evidence due to recent evidence and Food and Drug Administration (FDA) action that highlight safety concerns and potential overuse of ESAs for this population. This commenter also supported CMS’ commission of a TA from an outside entity and suggested we not move forward on a proposed decision until the conclusion of this TA.
A few commenters mentioned that the FDA’s Cardiovascular and Renal Drugs Advisory Committee (CRDAC) will be tentatively meeting this Fall [2010] to review the full range of evidence on ESA benefits and risks. These commenters suggested that the CMS not move forward on a proposed decision until after this meeting takes place.
Several commenters provided literature citations and/or other materials with comments. Full text comments without personal health information can be viewed at: https://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=245&ExpandComments=n&ver=6&NcaName=Erythropoiesis+Stimulating+Agents+(ESAs)+for+Treatment+of+Anemia+in+Adults+with+CKD+Including+Patients+on+Dialysis+and+Patients+not+on+Dialysis&bc=BEAAAAAAEAAA&.
Proposed Decision Comment Period: 3/16/11-4/15/11
During this comment period, CMS received a total of 21 comments. Of these public comments, six were from patient advocacy groups, five were from professional societies, four were from physicians, two were from research groups (one being the requestor), two were from pharmaceutical sponsors of ESAs, and two were from individuals whose professional background or affiliation could not be ascertained. Some of the comments included citations of abstracts, textbooks, and published articles. The two pharmaceutical sponsors provided extensive white papers/briefing documents with unpublished, proprietary data not vetted by the FDA. No individual/group providing comments provided copies of published articles in support of their statements. The cited published literature was reviewed and included in the final bibliography.
Of the 21 comments, 19 (90%) concurred with the proposed decision; two (10%) did not agree with the proposed decision. The comments addressed 13 aspects of the PDM and the decision process. Both the commenters who concurred with the decision and those who did not, frequently cited one or more of these 13 topics as justification for their conclusions about the decision. The topics addressed include: the assessment of the evidence by CMS and technology assessments performed for CMS; the role of the Food and Drug Administration (FDA) and its label; the roles of the Erythropoiesis Stimulating Agent Policy (EMP), Prospective Payment Program (PPS), and Quality Improvement (QIP); the role of specific guidelines as evidence; the use of ESAs for quality-of-life; transfusion reduction and transplantation outcomes; the role of ESA dose response in safety; the need for comparative effectiveness data; physician autonomy; and Medicare costs and reimbursement. For efficient organization and ease of reading, we have grouped the comments and their respective responses by theme.
1. Assessment of the Evidence
Several of those providing comments stated that while they concurred with the decision (5 of these 19 and 24% of the total number of commenters); they disagreed with CMS’s assessment of the evidence, the questions for the Transfusion-Transplantation technology assessment, and/or the Transfusion-Transplantation technology assessment analysis product. One of the pharmaceutical sponsors requested that the CMS review and analysis be changed based on comments from “stakeholders” and “authoritative bodies”.
Two of those providing comments stated that they did not concur with the decision, but concurred with CMS’s assessment of the evidence.
Response: CMS itself conducted an extensive review of the literature. An independent literature search conducted by the U.S. Agency for Healthcare and Research Quality (AHRQ), confirmed the completeness of the CMS dataset. No person/group providing comments submitted a published randomized clinical trial that had not been reviewed by CMS.
CMS notes that basic data such as dose response are not available, that cited pivotal registration studies remain unpublished, and that CMS has been asked to consider incomplete and non-vetted data in white papers and briefing documents. As indicated in the MEDCAC meeting, CMS also performed exploratory literature searches to determine what kinds of questions might be answered with a technology assessment. CMS agrees that there are fundamental questions about transfusion, sensitization, and transplant graft survival risk that remain unanswered, but notes that a technology assessment cannot provide definitive answers to such questions in the absence of clinical studies. No person/group providing comments at the MEDCAC meeting, in response to the draft technology assessment document, or in response the proposed coverage decision submitted substantive data for review. (Technology Assessment Report: The Impact of Pre-Transplant Red Blood Cell Transfusions in Renal Allograft Rejection, Disposition of Comments, Project ID: RENT0610)
As such, CMS believes its review and assessment of the evidence is accurate.
2. Food and Drug Administration (FDA)
Several commenters who concurred with the decision (eight of these 19 and 38% of the total number of commenters) indicated that the clinical practice is consistent with the FDA label, that the FDA should be the final arbiter, and/or that the latest FDA panel did not support changes to the label.
Two of those providing comments who did not concur with the decision (two of these two or 10% of the total number of commenters) cited changes to the FDA label addressing thrombo-embolic events, cardiovascular disease, and mortality. One of those providing comments who did not concur with the decision also cited the safety concerns expressed by FDA staff using quotes from the October 2010 CRDAC meeting during with the TREAT data were discussed.
Response: CMS and FDA share a common interest in improving the health of patients through the availability of safe, effective, and affordable medical products, yet CMS and the FDA have different mission statements and different statutory authority. CMS is tasked with serving the interests of the Medicare beneficiary populations. One of the ways in which it does so is through coverage policy decisions that determine what is “reasonable and necessary”. Data on “safety and efficacy” from the FDA constitute one of the elements used by CMS to inform its coverage decision process.
The FDA label is a negotiated legal document between the pharmaceutical sponsor and the FDA. The FDA may request specific study designs and endpoints, but can review only those materials which are submitted for initial approval or during the life cycle of a drug. If the data are thought to be sufficient to support an indicated use, a descriptive label is crafted. Labels may change with the discovery of new data or a new understanding of the scientific context of older data. This is evidenced by the serial changes including “BOXED warnings” in the FDA labels for ESAs since 2004. (See FDA section in NCD.)
The FDA label does not stipulate a specific hemoglobin-hematocrit level, in part, because, prior to the initiation of studies, the FDA did not establish the level of anemia for which mitigation was physiologically required. Furthermore, the FDA did not receive studies in which patients stratified on the basis of ESA-naïve hemoglobin (hematocrit) levels were given fixed ESA doses and the dose response and clinical responses assessed (except for the first eight weeks of the U.S. pre-dialysis study conducted by Teehan at al. 1991). The FDA did receive multiple studies in which patients were randomized to titration with escalating ESAs doses in an attempt to achieve pre-specified hemoglobin (hematocrit) goals. Many subjects did not achieve the pre-specified hemoglobin (hematocrit) goals. Mortality/cardiovascular morbidity appeared to be greater in patients who were unable to respond to ESAs and reach hemoglobin (hematocrit) goals. Because the study design: a) used a limited number of comparative hemoglobin (hematocrit) targets; and b) created confounding between hemoglobin (hematocrit) and dose, the determination of a “specific safe level” for hemoglobin (hematocrit) was not possible. In addition, although such studies did not generate sufficient dose information, the FDA ESA labels do include cautionary information about patients who respond poorly to dose escalation. (See Hypo-responder section in FDA label and the ESA Resistance sections in the NCD and Comments section.)
Like the MEDCAC for CMS, the FDA’s advisory committee (AC) panels such as the Cardio-Renal Advisory Committee (CRDAC) act in an advisory capacity to FDA. The panel's advice is considered within the context of the questions posed to the panel and may not be generalizable. Also, additional considerations/data available to FDA but not presented to the advisory committee, may also contribute to the FDA final determinations. A careful review of the transcripts for the 2010 CMS MEDCAC meeting and the 2010 FDA CRDAC meeting revealed the almost universal concern by both sets of panelists regarding ESA safety, but an inability to translate that concern into a specific voting recommendation because the comparative study designs utilized since the late 1980s did not/could not provide the type of information required. (CMS March 2010 MEDCAC meeting; FDA October 2010 CRDAC meeting)
CMS does note that while several commenters cited the importance of adherence to the FDA label, they were restrictive in their adherence. They cited hemoglobin targets, but supported ESA use for quality-of-life (QoL) and transfusion reduction despite the removal of the quality-of-life indication and the quality-of-life benefit statement from the label and the absence of "transfusion reduction" within the indication section in the labels for darbepoetin or pegylated erythropoietin. (See the QoL sections in the NCD and Comment sections.)
3. Quality Improvement Program (QIP)
Several of those providing comments (eight of 21; 38%) cited the measures employed by the QIP as evidence for coverage criteria and/or have indicated that coverage policy should be consistent with the QIP measures.
Response: The QIP was established by the Medicare Improvements for Patients and Providers Act (MIPPA ; Public Law 110-275) in 2008 as part of a prospective payment system (PPS). (See www.federalregister.gov/articles/2011/01/05/2010-33143/medicare-program-end-stage-renal-disease-quality-incentive-program .) Effective January 1, 2012, providers/facilities are required to meet a performance score for specified quality measures to obtain maximum payment. While the QIP is outside the scope of the NCD, the Agency is reviewing all of its related policies to ensure that there is no conflict.
4. Erythropoietin Monitoring Policy (EMP)
Several of those providing comments who concur with the decision (five of these 19 and 24% of the total number of commenters) have indicated that coverage policy should be consistent with the EMP and/or that the EMP reduces ESA use.
One the commenters who did not concur with the decision noted that the EMP permits CMS payments for ESA claims with hemoglobin (hematocrit) levels in excess of the hemoglobin (hematocrit) targets delineated in the FDA ESA labels (at the time of the closure of the comment period). The commenting group also noted that current CMS policy which encourages/permits high ESA dosing endangers public health and is a wasteful expenditure of Medicare resources.
Response: The EMP is a claims processing mechanism applied to ESAs provided as an ESRD benefit under §1881(b) of the Social Security Act. The EMP, which was updated in 2008, limited payment on billing claims when the hematocrit (hemoglobin) exceeded 39% (13 g/dl). It also established new medically unlikely edits (MUE) under which claims for erythropoietin in excess of 400,000 U and darbepoetin claims in excess of 1200 ug were returned for presumed errors. The EMP is not a coverage policy. Further discussion of the EMP is outside the scope of the NCD.
5. Prospective Payment System (PPS)
Several of those providing comments (four of 21; 19%) indicated that the PPS will reduce ESA use, and, as such, a coverage policy is not warranted.
Response: The PPS is a bundled payment system which is separate from the coverage decision process. In the Medicare Improvement for Patients and Providers Act (MIPPA; Public Law 110-275) of 2008, the HHS Secretary was instructed to implement by January 1, 2011 a payment system under which a single payment is made under title XVIII to a provider of services or a renal dialysis facility for renal dialysis services. The payment would cover costs of the dialysis treatment and certain routine drugs, laboratory tests, and supplies furnished at home or in a facility. (See www.cms.gov/ESRD Payment/.) (Accessed May 16, 2011) Further discussion of the PPS and the QIP performance measures used by the PPS is outside the scope of the NCD.
6. Kidney Disease Outcomes Quality Initiative (KDOQI™) Guidelines from the National Kidney Foundation (NKF)
Several of those providing comments (4 of 21; 19%) cited the KDOQI guidelines as evidence.
Response: CMS reviewed all of the available relevant guidelines. (See Guidelines section in NCD.) The KDOQI guidelines were one of these many guidelines. CMS does note that types of professional society statements may differ in their rigor, e.g., position statements versus guidelines. Conclusions may be based on consensus or they may be based on a learned approach with a complete literature search, criteria for grading the evidence, and conclusions based on the quality and reproducibility of the studies. CMS conducted its own review of the literature, some of which may have formed the evidentiary basis for the many guidelines.
The latest KDOQI guidelines were developed in 2006 and updated in 2007. The guidelines included studies that were underpowered (i.e., too short or too small) for assessment of mortality and cardiovascular events, but nonetheless categorized as grade “A” quality studies: Canadian Erythropoietin 1990-1991 [n=118], Foley 2000 [n=146], Parfrey 2005 [n=596], Levin 2005 [n=172], Revicki 1994-Roth 1995 [n=83], Ritz 2007 [n=172], Roger 2004 [n=155]). The guidelines do not include the latest information such as that provided by the TREAT study (Pfeffer 2009 and its post hoc analysis, Solomon 2010) and that which served as the impetus for the ongoing Clinical Evaluation of the Dose of Erythropoietins Trial (CEDOSE) (NCT00827021; principal investigators: Giovanni and Paolo Strippoli) funded by the Italian government and Consorzio Mario Negri Sud. (See http://clinicaltrials.gov/ct2/show/NCT00827021 .) In addition, all guidelines which address ESA use carry this notation from the U.S. Agency for Healthcare and Research Quality (AHRQ) National Guideline Clearinghouse: This guideline references a drug(s) for which important revised regulatory and/or warning information has been released. February 16, 2010 - Erythropoiesis-Stimulating Agents (ESAs): Procrit, Epogen and Aranesp: The U.S. Food and Drug Administration (FDA) and Amgen notified healthcare professionals and patients that all erythropoiesis-stimulating agents (ESAs) must be used under a risk evaluation and mitigation strategy (REMS) risk management program. As part of the risk management program, a Medication Guide explaining the risks and benefits of ESAs must be provided to all patients receiving an ESA . (See www.guideline.gov/content.aspx?id=11651&search=erythropoietin+ and+kidney.) (Accessed May 16, 2011)
7. Quality-of-Life (QoL)
Several of those providing comments and who concurred with the decision (14 of these 19 and 67% of the total number of commenters) stated that quality-of-life is an important indication for ESA use. One of those providing comments indicated that quality-of-life could be difficult to assess. Six cited primary studies or reviews of quality-of-life studies.
One of those who did not concur with the decision (one of these two and 5% of the total number of commenters) indicated that there was no evidence for a quality-of-life benefit.
Response: Although quality-of-life issues may be a consideration in coverage decisions, no one has provided CMS with prospective, controlled,clinical trial data that demonstrated sustained improvement in clinically relevant quality-of-life parameters of sufficient magnitude to be clinically important and that correlated to an interval change in hemoglobin (hematocrit). The studies provided to CMS included primary studies or reviews of primary studies that were uncontrolled, unblinded, small, too short to establish durability, and/or used measurement tools that had not been validated in general or not validated in the specific population being studied.
CMS notes that the FDA released draft guidance for patient-reported outcomes (PRO) and quality-of-life claims in 2006. The evidentiary criteria for claims of health-related quality-of-life for ESAs were publicly discussed at an FDA advisory committee meeting in 2007 with sponsors taking an active role. (Amgen/OrthoBiotech Briefing Document September 2007; FDA Trentacosti Slides) The statements in the ESA labels asserting improvements in quality-of-life for ESAs were subsequently removed from labeling. The final PRO guidance was released by the FDA in 2009 (FDA PRO Guidance). No one providing comments, including sponsors, acknowledged or addressed the removal of the quality-of-life statements from the FDA label. No one providing comments provided clinical trial evidence that met the PRO criteria established by the FDA or other competent source.
8. Transfusion
Several commenters who concurred with the decision (six of these 19 and 29% of the total number of commenters) noted that adverse events were associated with transfusions. Several of those providing comments and concurring with the decision (13 of these 19 and 62% of the total number of commenters) stated that restricted ESA use would result in more transfusions and more sensitization and decreased likelihood of receiving a renal transplant. Of these, one stated that anemia is an unmet medical need in African-Americans and women; another stated that African-Americans require higher ESA doses for anemia management, and yet another stated that African-Americans were more likely to become sensitized presumably because they are at risk of receiving more transfusions.
Two of those who concurred with the decision noted limitations in data for transfusion reduction by ESAs. One stated that the transfusion data were flawed, but that secular trend data provided indirect proof of efficacy. Another stated that it would be unethical to conduct a) placebo-controlled studies to assess transfusion requirements at various hemoglobin levels and b) active-control trials with ESAs or transfusions at various hemoglobin levels despite the acknowledged evidence gaps.
Two of those who did not concur with the decision noted the insufficient data for transfusion reduction claims.
One of the pharmaceutical sponsors stated that ESA therapy should be used at doses no higher than that necessary to achieve and maintain hemoglobin at a level sufficient to avoid transfusion.
Response: Jerry Holmberg, Ph.D., Senior Advisor for Blood Policy in the U.S. Department of Health and Human Services (HHS), Executive Secretary for the HHS Advisory Committee on Blood Safety and Availability, and recipient of the American Association of Blood Bank’s (AABB) President’s Award for his work in biovigilance, presented a synopsis of the adverse events associated with transfusions and their relative decline over time at the March 2010 MEDCAC meeting. (See MEDCAC meeting slide set and transcripts.)
The evidence for transfusion reduction by ESAs is limited. There were three, small, controlled pivotal registration studies for erythropoietin: the Canadian Trial in dialysis patients (N=118) with some reduction of transfusion in the high hemoglobin target group, study 8701 in hemodialysis patients (N=68), and the US Recombinant Human Erythropoietin Predialysis Study (N=117) in which there were no transfusions. (Canadian Erythropoietin Study Group 1990, Teehan 1991) (See tables 4 and 5 in the Pivotal Registration Studies section of the NCD.) Study 8701 has never been published and the FDA does not have its primary reviews or copies of the sponsor’s dataset/clinical report accompanied by the required attesting signatures. (MEDCAC members made inquiries regarding these data. See 1/19/2011 MEDCAC meeting transcripts.) The pivotal registration studies for darbepoetin and pegylated erythropoietin did not provide information on transfusion reduction because they employed a non-inferiority design and an active (ESA) control.
Furthermore, the evidence for transfusion reduction by ESAs is flawed because no studies (pivotal registration studies or post-registration studies) used validated criteria for hemoglobin level at which a transfusion should be performed (Eckardt 2001), studies did not have an the established protocols for transfusion, studies did not utilize validated criteria for hemoglobin assessment during the hemodialysis cycle (Bellizzi 2002, Movilli 2002, 2004), and studies did not collect information regarding the indications for transfusion (chronic anemia versus medical-surgical procedure versus uremic bleeding diathesis). The absence of a blind, in addition to a validated transfusion treatment algorithm, limited the transfusion information that can be derived from major post-registration studies such as NCHT, CREATE, and CHOIR.
It is possible that African Americans have been treated inappropriately for presumptive anemia despite the fact that it has long been known that hemoglobin concentrations in healthy African Americans are normally 0.5 to 0.73 g/dl lower than hemoglobin levels in Caucasians. ( Dallman 1979, Jackson 1990, NHANES II 1982, Robins 2004, 2007, Williams 1981) Historical data have suggested that renal transplant outcomes are worse in African Americans, but the underlying etiology(ies) has(have) not been firmly established.(Gordon 2010) Recent data suggest that renal graft and patient survival at one, three, and five years was similar for highly sensitized (PRA >80%) African Americans and non-African Americans receiving virtually cross-matched deceased donor organs, induction therapy with basiliximab and maintenance therapy with tacrolimus, mycophenolate acid, and glucocorticoids. (Ren 2011)
9. Transplantation
Several of those providing comments (13 of 21 or 62%) stated that restricted ESA use would result in more transfusions and more sensitization and decreased likelihood of receiving a renal transplant. Two of these indicated that renal transplants are the best treatment for ESRD. Another stated that United Network for Organ Sharing (UNOS) and U. S. Renal Data Systems (USRDS; CMS data compiled by a contractor for the National Institutes of Health [NIH]) should have been included in renal transplant discussions.
Response: The speakers at the January MEDCAC meeting included Dr. James Bowman, a renal transplant surgeon at the U.S. Health Services Research Administration (HRSA), who works with the UNOS program. Publically available UNOS and USRDS data were utilized by several of the speakers. Factors that could impact transplantation eligibility such as age, co-morbid conditions, and transfusions for medical-surgical procedures or uremic bleeding were addressed.
The evidence for transfusion reduction by ESAs is limited and flawed. (See NCD and Comment sections for Transfusion.) The link between transfusions, transfusion reduction by ESAs for chronic anemia versus uremic bleeding and medical-surgical procedures, the various causes of organ rejection, and clinical outcomes is not easily quantifiable with the available data.
The assessment of sensitization as measured by panel reactive antibodies (PRA) have been complicated by a lack of standardization in assays, changes in assays over time, and factors contributing to intra-patient variability in test results. The calculated PRA (cPRA) value provides more uniformity. Specific problematic antigens are identified so that appropriate organs are offered and inappropriate organs are not offered.
Highly sensitized patients are thought to be at the highest risk for transplant rejection/poor graft function. Highly sensitized patients have been defined as those with cPRA ≥80%. Between 5 and 10% of patients on the renal transplant waitlist have cPRA scores ≥80%. (Figure 10; USRDS 2010 Atlas, p 322.) With advances in immune suppression, organ preservation, and patient management, more restrictive cPRA criteria for being designated highly sensitized are currently under discussion. (See OPTN/UNOS Histocompatibility 7/7/2010 and 1/19/2011 Draft Meeting Summaries, 7/7/2010 Goals, and 7/7/2010 Proposed Sliding Scale Criteria).
Figure 10 Waitlist Duration by PRA Value
Figure 11 Waitlist Duration by Transfusion Status
Most blood transfusions for renal patients are administered in the in-patient setting, and not in dialysis centers. Although there are sections in hospital claims for transfusions (e.g. National Claims History (NCH) blood pints furnished, NCH blood pints replaced, NCH blood pints not replaced, and NCH blood deductible pints) and revenue center trailers (e.g. 0380 blood general, 0381 packed red cells, and 0382 whole blood), payments are made on the basis of Diagnosis-Related Groups. In the absence of an incentive to complete these data fields, under-reporting bias complicate data derived from these sources. The U.S. Health Services Research Administration (HRSA) collects transplant data through the Organ Procurement Transplantation Network (OPTN) and the United Network for Organ Sharing (UNOS) and contracts with the Scientific Registry of Transplant Recipients (SRTR) to analyze the data. Currently HRSA data collection for transfusions is limited to a single question “Did patient receive any pre-transplant blood transfusions?” with the response “Yes, No, or Unknown” in the Kidney Transplant Recipient Registration Worksheet (and not the Kidney Transplant Candidate Registration Worksheet). As such, we do not have good data collection systems in place to obtain contemporaneous data on transfusions (e.g., number of transfusions, number of units transfused, hemoglobin at time of transfusion, and reason for transfusion) making it difficult to make risk assessments about the impact of transfusion (and not the co-morbidities contributing to the need for transfusion) on sensitization and transplant graft survival. (See transcripts for the 1/19/2011 MEDCAC.)
The available data suggest that the median waitlist time for those who have received one or more blood transfusions is only one to three months longer than those who have not been transfused. (Figure 11; USRDS 2010 Atlas, p 322.) (Speakers at the January MEDCAC meeting did note that an increasing number of CKD patients are not candidates for transplantation and that the underlying cause is age and/or co-morbid disease. Currently the “active” wait-list is ~55,570 patients, but there is an “inactive” waitlist of ~33,180 patients in the setting of 16,898 kidney and 828 pancreas-kidney transplants in 2010.) (See Delmonico 2008 and the UNOS Weekly Fact Sheet May 2011.)
10. ESA Resistance
Several commenters concurring with the decision (three of these 19 or 14% of the total number of commenters) indicated that ESA resistance may be an important safety parameter. One noted that it may be more important than hemoglobin level.
Response: CMS concurs that resistance to ESAs is a marker for or a cause of poor outcomes. CMS notes five studies which were conducted in a variety of populations and/or using different study designs, but whose conclusions were similar.
1—The BEST study was performed in breast cancer patients.(Leyland-Jones 2003, 2005) In figure 4 of the paper, one observes that exposure to an ESA resulted in a greater mean hemoglobin and a greater likelihood of death, but that the hemoglobin levels were lower in patients who died versus those who did not die.
2—The observational study by Zhang used USRDS data from dialysis patients. (Zhang 2004) In figure 2 of the paper, one observes that the likelihood of death at one year was increased when the level of anemia at t=0 was more severe or the erythropoietin dose at t=0 more elevated. If one was anemic despite being on high doses at t=0, the likelihood of death was approximately 2.5 times that found in non-anemic and low erythropoietin dose patients.
3—The NCHT was performed in dialysis patients. Patients were randomized to high or low hemoglobin targets. The Kilpatrick paper is a post hoc analysis of the NCHT. (Kilpatrick 2008) In figure 2 and table 2 of the paper, one observes that those in the lowest quartile for hematocrit response to erythropoietin had a death rate approximately 2.5 times that observed in the highest response quartile despite similar hematocrit levels at baseline.
4—The CHOIR study was performed in pre-dialysis CKD patients. The Szczech paper is a post hoc analysis. (Szczech 2008) In table 2, one observes that patients who were unable to achieve target hemoglobin values by either four months or nine months experienced more cardiovascular events or mortality and were more likely to be on higher doses of erythropoietin. Within each treatment arm (higher hemoglobin target versus lower hemoglobin target), patients on the highest doses had more cardiovascular events or mortality than those on lower doses.
5 The TREAT study was performed in pre-dialysis CKD patients. The Solomon paper is a post hoc analysis. (Solomon 2010) In table 1, one observes that the first quartile had the poorest hematologic response to darbepoetin in the first 12 weeks of treatment. The response was not predicted by baseline hemoglobin. In table 2, one observes a higher risk of death and/or a cardiovascular endpoint if one had a poor initial hemoglobin response to darbepoetin.
These analyses suggest that high hemoglobin levels were not the cause of these particular adverse outcomes (although it is possible that they play a role in other adverse events). (See hypo-responder section of the FDA label.) (See ESA Resistance section of the NCD.)
11. Autonomy
Several of those providing comments (nine of 21 or 43%) stated that the physician should have autonomy to make decisions regarding ESA. Three of those commenting stated that there should be no coverage restrictions unless there is definitive proof of harm. Two stated that CMS should preclude local contractors from preparing LCDs.
Response: Section 1862(a)(1)(A) of the Social Security Act says that CMS must not pay an item or service unless it is found to be “reasonable and necessary” for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member except for select items and services including some preventive services (section 1395x(ddd)(1) of this title).
In the absence of a National Coverage Determination (NCD) and other statutory requirements, coverage is at the discretion of the local Medicare contractor. (Section 1869(f) (2) (B) of Social Security Act.)
12. Medicare Expenditures
One of those providing comments who concurred with the decision stated that financial considerations should not influence physician decisions regarding choice of therapy. Another stated that he believed that restrictions on ESAs would contribute to complications and health care costs. Another stated that small facilities would be faced with financial non-viability versus sub-optimal patient care if a more restrictive NCD was implemented.
Two commenters who did not concur with the decision stated that the lack of a more restrictive NCD was fiscally imprudent given the lack of evidence for health benefits and the presence of evidence of harm. One of these stated that ESA use should be restricted to the lowest dose to minimize the need for blood transfusions, the only clinical benefit that their group considered to have an evidentiary basis.
Response: Coverage decisions are made on the basis of “reasonable and necessary.” (See “reasonable and necessary” definition under the Autonomy section.) Cost or reimbursement is not part of an NCD and is outside the scope of the NCD.
13. Comparative Effectiveness
One of those providing comments who concurred with the decision requested that CMS conduct comparative effectiveness research.
Response: In general, Medicare has not conducted or utilized comparative effectiveness research. The U.S. Agency for Healthcare and Research Quality (AHRQ ) was authorized by section 1013 of the Medicare Modernization Act (MMA) of 2003 authorized AHRQ to conduct and support research with a focus on “outcomes, comparative clinical effectiveness, and appropriateness of health care items and services” for Medicare and Medicaid enrollees. (CBO 2007) Additional funding for this program was provided through the American Recovery and Reinvestment Act of 2009. The Congress through the 2010 Patient Protection and Affordable Care Act established the Patient-Centered Outcomes Research Institute (PCORI) an independent, non-profit organization to conduct research to determine the effectiveness, benefits and harms of different diagnostic modalities and treatment options, e.g., drugs, medical devices, and surgical procedures, as well as different health care delivery systems for different patients.
Summary Response
The ecology of ESA prescribing is complex. It is obvious that the clinical science has evolved since ESAs were first approved for marketing in the United States many years ago. We are in the midst of a transition from a well intentioned but in hindsight naïve assumption that anemia and its management were well understood scientifically. That overly simple approach supported an ESA-centric response to low hemoglobin laboratory values that is only recently being challenged by a growing body of evidence.
Medicare and its programs are an important aspect of that ecosystem. CMS has had policy and payment avenues that have historically used payment and reporting incentives or disincentives to better align Medicare payment with the evolving evidence and we expect that the implementation of these programs will continue to evolve with the evidence.
At this point we believe that we should, rather than publishing a National Coverage Determination, monitor the use of ESAs in light of the recent implementation of ESRD bundled payment and consider how we can use all of our policy avenues in an integrated response to ESA overuse.
VIII. CMS Analysis
A. Analysis Framework
National coverage determinations (NCDs) are determinations by the Secretary with respect to whether or not a particular item or service is covered nationally by Medicare (§1869(f)(1)(B) of the Act). In order to be covered by Medicare, an item or service must fall within one or more benefit categories contained within Part A or Part B, and must not be otherwise excluded from coverage. Moreover, with limited exceptions the expenses incurred for items or services must be “reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member.” See §1862(a) (1) (A) of the Act. This section presents the agency’s evaluation of the evidence considered and conclusions reached for the assessment
B. Analysis
Questions:
A. Is the evidence sufficient to conclude that the underlying cause for anemia in Medicare beneficiaries who have renal disease and are not on dialysis is absolute and irreversible erythropoietin deficiency?
B. If the answer to question A is affirmative, is the evidence sufficient to conclude that erythropoiesis (erythrocyte) stimulating agent (ESA) therapy affects health outcomes (including survival, cardiovascular event rates, exercise capacity, progression of renal disease, quality-of-life, transfusion rates, and ability to receive a transplant) when used by Medicare beneficiaries who have renal disease and are not on dialysis?
C. If the answer to Question B is affirmative, is there sufficient evidence to determine which characteristics of the patient, the disease, or the treatment regimen reliably predict a favorable or unfavorable health outcome when used by Medicare beneficiaries who have renal disease and are not on dialysis?
D. Is the evidence sufficient to conclude that the underlying cause for anemia in Medicare beneficiaries who have renal disease and are on dialysis is absolute and irreversible erythropoietin deficiency?
E. If the answer to question D is affirmative, is the evidence sufficient to conclude that erythropoiesis (erythrocyte) stimulating agent (ESA) therapy affects health outcomes (including survival, cardiovascular event rates, exercise capacity, quality of life, transfusion rates, and ability to receive a transplant) when used by Medicare beneficiaries who have renal disease and are on dialysis?
F. If the answer to Question E is affirmative, is there sufficient evidence to determine which characteristics of the patient, the disease, or the treatment regimen reliably predict a favorable or unfavorable health outcome when used by Medicare beneficiaries who have renal disease and are on dialysis?
In seeking to address these questions, CMS sought evidence on the underlying clinical science
- For anemia in general, what are the physiologic criteria for intervention?
- What are the causes for anemia in renal disease?
- Do the causes differ along the spectrum of renal dysfunction?
- When is anemia erythropoietin-mediated?
- Are there validated criteria for transfusion?
- For what reasons do renal patients receive transfusions?
- Is there evidence that ESAs eliminate/reduce the need for transfusion when validated criteria for transfusion are employed?
-
- Is there evidence of improved clinical outcomes from ESA therapy?
- Is there evidence of potential harm from ESA therapy?
- Do we have sufficient data to determine whether the hemoglobin level or ESA dose contributes to benefit or harm?
- How do ESA dose levels in the U.S. compare to dose levels elsewhere?
- If ESA resistance, i.e. requirement of more than physiologic replacement, is present, is there evidence that patient outcomes are improved by continued/increased ESA dosing?
Our analysis is made more complex because of certain historical assumptions that have been recently challenged. In the late 1980s, erythropoietin was developed to treat anemia and reduce the need for transfusions, especially with the advent of AIDS and a limited ability to screen the blood supply. (OTA 1985, OTA 1990) It was presumed that many complications of renal disease were related to anemia rather than to the underlying disease or comorbidities. The renal failure population was relatively small, thus the orphan drug designation, and homogenous. (Coster 1992, OTA 1990)
CMS has carefully reviewed the historical context of renal disease, anemia management, secular changes in the renal patient population, and an evolving ESA database in which hypothesized benefits have been assessed more rigorously.
1. Physiology and Hemoglobin Requirements
Hemoglobin level requirements for physiologic function remain poorly understood. Most of the observational and interventional data are from the acute care setting. In one of the few controlled studies, Hebert found that 30 day mortality rates were not improved by liberal transfusion policies (transfusion for hemoglobin levels less 10 g/dl and maintenance with hemoglobin levels between 10 and 12 g/dl) compared to restrictive transfusion policies (transfusion for hemoglobin levels less 7g/dl and maintenance with hemoglobin levels between 7 and 9 g/dl) in 838 euvolemic, anemic, critically ill intensive care patients stratified by disease severity (APACHE II score). (Hebert 1999) Mortality was actually increased in relatively healthy (APACHE II score < 20) and young (< 55 years) patients. Results did not differ by patient subgroup: cardiovascular disease (n = 357) (Hebert 2001a), head injury (n = 67) (McIntyre 2006), mechanically ventilated (n = 713) (Hebert 2001b), and trauma (n = 204) (McIntyre 2004). In the same way, Hajjar et al. found that 502 cardiac surgery patients transfused to maintain a hematocrit of 30% or higher versus a hematocrit of 24% or higher had equivalent 30 day morality rates and severe morbidity (cardiogenic shock, acute respiratory distress syndrome, or acute renal injury requiring dialysis or hemofiltration). In the same way, Carson et al. found that 1007 hip-fracture surgical patients with known cardiovascular disease who transfused to hemoglobin levels in excess of 10 g/dl did not have more exercise tolerance at 60 days post-operation than the 1009 restrictive arm patients who were transfused after hemoglobin levels dropped to below 8 g/dl or patients became symptomatic. (Carson 2009-abstract, 2011 MEDCAC presentation, 2011)
Hemoglobin requirements in the chronic setting cannot be directly extrapolated from findings in the acute care setting because there are compensatory such as increases in 2,3 diphosphoglycerate which result in better oxygenation at the tissue level than what would otherwise be expected for a given plasma hemoglobin level. (Aberman 1985, Eckhardt 2001, Metivier 2000, McDonald 1977) There are no equivalent randomized trial data to assess the physiologic requirements of renal patients, either pre-dialysis or dialysis treated. The series of papers by the Collins group are frequently cited as the reason for achieving hemoglobin levels between 10 and 12 or higher “After adjusting for these confounding patient characteristics, our results showed that patients with hematocrit levels < 30% have significantly higher risk of all-cause & cause-specific death, compared to patients with hematocrit levels of 30% to < 33%. …After adjusting for severity of disease, the impact of hct levels in the 33% to <36% range becomes vulnerable to the number of patients included but still demonstrates a further 4% reduced risk of death. Overall, our findings suggest that sustained increases in hematocrit levels are associated with improved patient survival.” These papers, however, do not describe either the natural history of renal disease (because most of the population was treated with an ESA) or the specific effects of an intervention (because the presence of an intervention, ESA, and the size of the intervention, ESA dose parameters, were not included in the analyses.) Indeed another author group who performed a similar analysis, Madore et al. specifically cautioned against the extrapolation of such observational data in the absence of correction for therapeutic interventions and co-morbid disease. (Madore 1997)
The most recent systematic reviews conducted by Cochrane (Carliss et al. 2009) and the European Blood Alliance (McClelland et al. 2010) suggest that anemia management with transfusions to maintain or achieve hemoglobin levels > 10 g/dl does not confer physiologic benefit and that anemia management with transfusions is not required until the normovolemic hemoglobin level is below 6 or 7 g/dl unless there is evidence of physiologic decompensation or circulatory risk.
2. Causes of Anemia
Patients with renal disease, especially older patients with co-morbid chronic disease conditions and occult marrow dysfunction, may have anemia for a variety of reasons. Anemia, however, cannot be attributed to renal disease until the GFR is < 30 ml/min/m2. Longitudinal data show that as renal function declines in the months prior to dialysis and hemoglobin levels decline, erythropoietin levels actually rise. After dialytic removal of uremic toxins, there is a compensatory increase in hemoglobin levels and decline in erythropoietin levels. These data show that anemia prior to dialysis is not intrinsically mediated by the hormone, erythropoietin. Rather, it generally can be attributed to uremia. In the 6-12 month period after the onset of dialysis, there is further loss of renal tissue in most patients and erythropoietin levels typically permanently decline. In patients with preserved functional tissue, such as those with polycystic kidney disease, residual erythropoietin hormone production may persist. Hormone deficiencies are typically managed by replacement dosing. Physiologic replacement doses approximate 150 U/kg/week or less in normal weight subjects. Higher dose requirements, or resistance, suggest the superimposition of other disease processes such as inflammation, infection, drug-induced marrow fibrosis, or drug-interaction. In many of the registration studies, other causes of anemia were not rigorously excluded. Indeed many studies used only calculated GFR and/or included pre-dialysis subjects whose anemia could not even be attributed to renal dysfunction (uremia) because of filtration values between 30 and 59 ml/min.
3. Transfusions
Although ESAs were developed to reduce transfusion dependence, transfusions can be given for a variety of reasons including chronic anemia management, blood loss due to hemodialysis, bleeding diatheses secondary to uremia, and surgical procedures for renal and non-renal conditions. Unfortunately, the data in support of this indication is poor. To establish this claim, it is necessary to have validated criteria for transfusion, study protocols for transfusion, and documented adherence to such protocols in patients with erythropoietin-mediated anemia. As delineated above, the criteria for anemia intervention are not well established. As such, it was not possible to prepare evidence-based protocols for anemia intervention. The randomized clinical trials did not include criteria for transfusion except “clinical indication.” Information on the number of transfusions, number of units transfused (per person transfused), and the reasons for transfusion were not reported. Little is known about the characteristics of patients who received transfusion. Many studies failed to rigorously exclude other causes of anemia. The absence of blinding further complicated interpretation of transfusion data.
In particular it should be noted that only erythropoietin carries the indication for transfusion reduction. The subsequent ESAs used non-inferiority or equivalence data in support of their drug registration applications. As such, they were active controlled, and not placebo controlled studies. There is only one published/publicly available placebo-controlled clinical trial in dialysis patients using an FDA approved product: the Canadian study with 118 patients. These patients were markedly different than the current dialysis population. They were more anemic. They were younger. Their underlying renal disease differed; diabetes was excluded. There is only one published/publicly available placebo controlled clinical trial in pre-dialysis patients using an FDA approved product: the Teehan study with 117 patients. No transfusions were administered during the trial. Because renal patients receive transfusions for reasons other than chronic anemia, ESAs may not be able to eliminate the need for transfusions. The reviewing FDA medical officer for an unpublished darbepoietin registration study in dialysis patients noted “The 27% transfusion rate in the ARANESP group in Study 211 is quite substantial… In any case, the data do not make the case that ARANESP decreased the need for RBC transfusions, given the directionally opposite trend.”
4. Other Hypothesized Clinical Benefits
Based on the existing record, ESAs do not clearly improve quality-of-life. Indeed, the FDA removed health related quality-of-life claims from the ESA labels after a public hearing in 2007. The sponsor cited evidence from four studies from the initial circa 1988 drug approval application (three small controlled studies (one published EP 86-004, two unpublished 8701 and 8904) and one uncontrolled study 8601). The FDA cited multiple design inadequacies including blinding, failure to prospectively address missing data, post-hoc analysis, and the absence of any correlation (changes in) hemoglobin or hematocrit levels and (changes in) anemia symptoms. The FDA cited deficiencies in the test instruments that were used including problems with content validity and post hoc selection of test items. More recent, larger, and longer studies (CHOIR, CREATE, NCHT, and TREAT) did not demonstrate any clinically significant, durable improvements in (health-related) quality-of-life using validated instruments.
Based on the existing record, ESAs do not clearly improve exercise tolerance. Many of the studies were relatively short in duration and small in size. The largest study powered for exercise (Furuland 2003; n = 416) could not be completed because many of the subjects could not perform the testing. Despite the near complete penetration of ESA use in the dialysis population, the ability of patients, especially older patients, to ambulate declines during their first year on dialysis. This coupled with studies that suggest that exercise training programs can improve physical function, suggest that exercise performance and fatigue are related to a variety of variables.
Based on the existing record, ESAs do not alter the rate of renal disease progression. The earliest studies used surrogate markers and attempted to estimate renal function decline using the calculated slope of creatinine clearance of GFR change measured by a variety of methods over variable periods of observation. Only three randomized studies (CHOIR, CREAT, and TREAT) collected data on progression, but were limited because progression to dialysis was not a primary endpoint and the baseline data were not rigorous assessments of renal function. Nonetheless, none of these studies showed that ESA use or randomization to a higher hemoglobin target group decreased the likelihood or onset of dialysis, and the CREATE study suggested increased renal function decline.
Based on the existing record, the largest and longest randomized studies of intermediate cardiac endpoints, primarily left ventricular mass, did not show improvement. In addition, more definitive studies with cardiovascular events and/or survival (NCHT, CHOIR, CREATE, TREAT, and PEARL) did not show improvement in the higher hemoglobin target treatment arms.
5. Adverse Clinical Outcomes
Some of the earliest studies demonstrated that exogenous erythropoietin could result in fluid retention, hypertension, and vascular thrombosis. Later studies in renal patients suggested that chronic ESA use could result in decreased survival, increased rates of cardiovascular events, and increased rates of thrombosis (arterial and venous) (Table 28). It has been postulated that renal patients with the vascular endothelial dysfunction and impaired vascular relaxation associated with atherosclerosis may lack compensatory mechanisms for adaptation to the increased blood viscosity found with ESA use. (Eckhardt 2001, Metivieu 2000) In the TREAT study, there were more deaths in patients with a prior history, but no known active cancer at study entry in the higher hemoglobin target arm. Some of these latter studies suggest harm in the higher hemoglobin target arm, whether or not the target was achieved. These data suggest that dose or other drug exposure parameter may be more important than hemoglobin level as a mediator of harm for some adverse events. This role for dose is supported by new retrospective studies by Seliger et al. and Zhang et al. (Seliger 2011, Zhang 2011) More definitive conclusions about the degree and nature of the harm cannot be made because of premature discontinuation of the prospective trials before statistical significance was reached and/or the high withdrawal rates and poor follow-up in addition to the confounding introduced by the study design elements such as the absence stratification by ESA-naïve hemoglobin levels and/or absence of fixed dosing. Most of the hundred of studies that have been conducted in thousands of patients since the introduction of ESAs 20 years ago have not been structured to address these fundamental questions.
Table 28: Mortality and Cardiovascular Events in Major Trials
6. Dosage
In the classic paradigm, physiologic replacement of a missing hormone should result in normalization of function. In the non-classic paradigm, a hormone is used at higher than physiologic levels because of hormone resistance or to supplement endogenous pathways to achieve supraphysiologic or accelerated physiologic responses. It is a well known phenomenon that hormones and related molecules will bind to higher occupancy sites first and lower occupancy (non-classical) sites later-depending on dose and hormonal milieu. Residence time may also be important. Non-classic actions or pleiotropic effects may occur-especially in the setting of high dosing. Toxicology studies in animals, however, appear to have been limited in scope and did not include carcinogenicity studies. (Erythropoietin information limited to that contained in the label.) No fixed dose studies with stratification for co-morbid conditions and ESA-naïve hemoglobin levels have been conducted. None of the primary papers discuss response rate by dose.
Despite these deficiencies in the data, it is clear that administered ESA doses have increased markedly over time in the U.S (Figure 8). ESA dosing in the U.S. exceeds physiologic replacement and is approximately twice that in Europe despite equivalent hemoglobin results, and hemoglobin levels that exceed physiologic requirements and known to confer any beneficial clinical outcome (Tables 29, 30, and 31). The doses of this hormone that are being given are by definition supraphysiologic—more than replacement—especially immediately after IV administration dose. (Figures 1 and 2) The reasons for these differences in dosing practice remain unclear.
Recent retrospective studies suggest that acute stroke is more frequent in renal insufficiency patients, especially those with concomitant cancer (Seliger 2011), and mortality is greater in end stage renal disease patients, especially those with diabetes, when using higher ESA doses (Zhang 2011 in press).
Table 29: Hemoglobin Level and Erythropoietin Dose in the 2003 ESAM Cross-sectional Survey on Anaemia Management (Jacob 2005)
Table 30: Hemoglobin Level and Erythropoietin Dose in the 2002-3 DOPPS Cross-sectional Survey in Hemodialysis Patients (Pisoni 2004)
Table 31: Hemoglobin Level and Erythropoietin Dose in the UK Renal Registry Surveys in 1997 and 2007 (Burton 2000, Richardson 2009)
ESA Resistance
As we noted above, in the classic paradigm, physiologic replacement of a missing hormone should result in normalization of function. Indeed many, albeit not all, patients with end-stage renal disease are deficient in erythropoietin because of damage to the renal parenchyma. Their anemia is secondary to and highly responsive to low doses of ESAs. In the non-classic paradigm, a hormone is used at higher than physiologic levels because of hormone resistance or to supplement endogenous pathways to achieve supraphysiologic or accelerated physiologic responses.
Poor drug response, i.e., resistance, suggests the presence of other clinical factors. Infection (frank or occult), inflammation (from a variety of causes including occult malignancy and adipose-related cytokines), impaired bone marrow reserve, dialysis adequacy, concomitant anemia from other causes (including iron deficiency and the anemia of chronic disease associated with type 2 diabetes mellitus), and drug products (interactions with endogenous erythropoietin or exogenous ESAs or ESA direct effects on the marrow or ESA drug-packaging induction of autoantibodies) have all been implicated in ESA resistance. Rossert et al. (OrthoBiotech) conducted a post hoc analysis in a subset of the ECAP study population and reported that greater body mass (BMI), older age, attribution of diabetes as the underlying cause of renal disease, anemia, and use of angiotensin-converting enzyme< (ACE) or angiotensin II receptor blocking (ARB) anti hypertensive drugs were associated with higher erythropoietin dose requirements although these variables did not account for all of the variability in erythropoietin dosing. (Rossert 2006, 2007)
Exploration of the underlying cause of ESA resistance is important for patient outcomes. Kilpatrick et al. (Amgen) conducted a post hoc analysis of 1-year mortality in dialysis and ESA responsiveness in NCHT dialysis patients with pre-study hematocrit levels of 30 ±3 vol%. (Kilpatrick, 2008) The authors defined erythropoietin response as the weekly hematocrit change/erythropoietin dose increase. The patients in the lowest response rate quartile had the highest mortality (Table 32).
Table 32: Erythropoietin Resistance and Mortality (NCHT)
Similar findings were found in pre-dialysis patients in post hoc analyses of the CHOIR and TREAT data sets. (Solomon 2010, Szczech 2008)
Unfortunately, none of the published studies or FDA reviews discuss hemoglobin response rate by dose after stratifying by ESA-naïve baseline hemoglobin level. None of the studies were designed to prospectively assess erythropoietin resistance and putative variables. Many of the exclusion criteria for registration studies specifically excluded patients with high ESA dosing requirements or risk factors for resistance. The pivotal studies for pegylated erythropoietin excluded patients with elevated C-reactive protein (CRP) levels. The Resistance to ErythroPoietin Effectiveness Trials (REPEAT) (ClinicalTrials.gov identifier: NCT00319150)(Principal investigator K E Yeates; Sponsor OrthoBiotech) which was initiated in 2006 was terminated. (www.clinicaltrials.gov/ct2/show/NCT00319150?term=yeates+and+erythropoietin&rank=1; accessed February 11, 2011). There have been no drug interaction studies for medications such as ACE inhibitors, which are frequently used in the renal and diabetic patient populations. There are no long-term studies with bone marrow biopsies (published in entirety) to assess drug-induced fibrosis although early unpublished toxicology data and more recent molecular biologic data have suggested this possibility.
Conclusion
ESAs are being used with supraphysiologic dosing at hemoglobin/hematocrit levels higher than those used to avoid transfusions. Despite an exhaustive search, we identified no high quality, randomized clinical trials that were of sufficient design, duration, and power to definitely determine that ESAs provided clinical benefits other than increasing hemoglobin, a putative intermediate clinical surrogate in patients with documented erythropoietin-mediated anemia. The available data do not support claims of ESA benefit for health-related quality of life, exercise performance, cardiac function, or survival. The evidence for transfusion reduction is limited because of the absence of validated criteria for transfusion, the absence of defined study protocols for transfusion, and the use of non-inferiority (or equivalence) study designs that lacked a placebo arm.
We identified no randomized clinical trials that used fixed doses and stratification by ESA-naïve hemoglobin levels to better define the response rate to physiologic dosing, assess dose-related safety, and exclude the confounding associated with hemoglobin levels and targets. We identified no good drug interaction studies. Despite the absence of complete publications in easily accessible medical journals, we did identify emerging evidence for harm including increased mortality, tumor progression, cardiovascular-thromboembolic events, and stroke in patients with renal insufficiency and/or renal failure. Although there are a plethora of studies comparing ESA preparations, dosing regimens and routes of administration, important fundamental data about ESA and their use are lacking. Optimal patient management dictates that patients with either primary (e.g. infection, occult cancer, dialysis inadequacy, or dysplasic marrow) or secondary (e.g., anti-erythropoietin antibody mediated pure red cell aplasia or drug-induced marrow fibrosis) ESA resistance be identified and the underlying causes addressed prior to dose increases. The current published studies are insufficient to delineate risk:benefit for the various patient populations, particularly the Medicare population.
IX. Final Decision
Given the totality of the currently available evidence, CMS will not issue a national coverage determination at this time for Erythropoiesis Stimulating Agents (ESAs) for Treatment of Anemia in Adults with CKD Including Patients on Dialysis and Patients not on Dialysis (CAG-00413N).
APPENDIX A
General Methodological Principles of Study Design
(Section VI of the Decision Memorandum)
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service is reasonable and necessary. The overall objective for the critical appraisal of the evidence is to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for patients.
We divide the assessment of clinical evidence into three stages: 1) the quality of the individual studies; 2) the generalizability of findings from individual studies to the Medicare population; and 3) overarching conclusions that can be drawn from the body of the evidence on the direction and magnitude of the intervention’s potential risks and benefits.
The methodological principles described below represent a broad discussion of the issues we consider when reviewing clinical evidence. However, it should be noted that each coverage determination has its unique methodological aspects.
Assessing Individual Studies
Methodologists have developed criteria to determine weaknesses and strengths of clinical research. Strength of evidence generally refers to: 1) the scientific validity underlying study findings regarding causal relationships between health care interventions and health outcomes; and 2) the reduction of bias. In general, some of the methodological attributes associated with stronger evidence include those listed below:
- Use of randomization (allocation of patients to either intervention or control group) in order to minimize bias.
- Use of contemporaneous control groups (rather than historical controls) in order to ensure comparability between the intervention and control groups.
- Prospective (rather than retrospective) studies to ensure a more thorough and systematical assessment of factors related to outcomes.
- Larger sample sizes in studies to demonstrate both statistically significant as well as clinically significant outcomes that can be extrapolated to the Medicare population. Sample size should be large enough to make chance an unlikely explanation for what was found.
- Masking (blinding) to ensure patients and investigators do not know to which group patients were assigned (intervention or control). This is important especially in subjective outcomes, such as pain or quality of life, where enthusiasm and psychological factors may lead to an improved perceived outcome by either the patient or assessor.
Regardless of whether the design of a study is a randomized controlled trial, a non-randomized controlled trial, a cohort study or a case-control study, the primary criterion for methodological strength or quality is the extent to which differences between intervention and control groups can be attributed to the intervention studied. This is known as internal validity. Various types of bias can undermine internal validity. These include:
- Different characteristics between patients participating and those theoretically eligible for study but not participating (selection bias).
- Co-interventions or provision of care apart from the intervention under evaluation (performance bias).
- Differential assessment of outcome (detection bias).
- Occurrence and reporting of patients who do not complete the study (attrition bias).
In principle, rankings of research design have been based on the ability of each study design category to minimize these biases. A randomized controlled trial minimizes systematic bias (in theory) by selecting a sample of participants from a particular population and allocating them randomly to the intervention and control groups. Thus, in general, randomized controlled studies have been typically assigned the greatest strength, followed by non-randomized clinical trials and controlled observational studies. The design, conduct and analysis of trials are important factors as well. For example, a well designed and conducted observational study with a large sample size may provide stronger evidence than a poorly designed and conducted randomized controlled trial with a small sample size. The following is a representative list of study designs (some of which have alternative names) ranked from most to least methodologically rigorous in their potential ability to minimize systematic bias:
Randomized controlled trials
Non-randomized controlled trials
Prospective cohort studies
Retrospective case control studies
Cross-sectional studies
Surveillance studies (e.g., using registries or surveys)
Consecutive case series
Single case reports
When there are merely associations but not causal relationships between a study’s variables and outcomes, it is important not to draw causal inferences. Confounding refers to independent variables that systematically vary with the causal variable. This distorts measurement of the outcome of interest because its effect size is mixed with the effects of other extraneous factors. For observational, and in some cases randomized controlled trials, the method in which confounding factors are handled (either through stratification or appropriate statistical modeling) are of particular concern. For example, in order to interpret and generalize conclusions to our population of Medicare patients, it may be necessary for studies to match or stratify their intervention and control groups by patient age or co-morbidities.
Methodological strength is, therefore, a multidimensional concept that relates to the design, implementation and analysis of a clinical study. In addition, thorough documentation of the conduct of the research, particularly study selection criteria, rate of attrition and process for data collection, is essential for CMS to adequately assess and consider the evidence.
Generalizability of Clinical Evidence to the Medicare Population
The applicability of the results of a study to other populations, settings, treatment regimens and outcomes assessed is known as external validity. Even well-designed and well-conducted trials may not supply the evidence needed if the results of a study are not applicable to the Medicare population. Evidence that provides accurate information about a population or setting not well represented in the Medicare program would be considered but would suffer from limited generalizability.
The extent to which the results of a trial are applicable to other circumstances is often a matter of judgment that depends on specific study characteristics, primarily the patient population studied (age, sex, severity of disease and presence of co-morbidities) and the care setting (primary to tertiary level of care, as well as the experience and specialization of the care provider). Additional relevant variables are treatment regimens (dosage, timing and route of administration), co-interventions or concomitant therapies, and type of outcome and length of follow-up.
The level of care and the experience of the providers in the study are other crucial elements in assessing a study’s external validity. Trial participants in an academic medical center may receive more or different attention than is typically available in non-tertiary settings. For example, an investigator’s lengthy and detailed explanations of the potential benefits of the intervention and/or the use of new equipment provided to the academic center by the study sponsor may raise doubts about the applicability of study findings to community practice.
Given the evidence available in the research literature, some degree of generalization about an intervention’s potential benefits and harms is invariably required in making coverage determinations for the Medicare population. Conditions that assist us in making reasonable generalizations are biologic plausibility, similarities between the populations studied and Medicare patients (age, sex, ethnicity and clinical presentation) and similarities of the intervention studied to those that would be routinely available in community practice.
A study’s selected outcomes are an important consideration in generalizing available clinical evidence to Medicare coverage determinations. One of the goals of our determination process is to assess health outcomes. These outcomes include resultant risks and benefits such as increased or decreased morbidity and mortality. In order to make this determination, it is often necessary to evaluate whether the strength of the evidence is adequate to draw conclusions about the direction and magnitude of each individual outcome relevant to the intervention under study. In addition, it is important that an intervention’s benefits are clinically significant and durable, rather than marginal or short-lived. Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits.
If key health outcomes have not been studied or the direction of clinical effect is inconclusive, we may also evaluate the strength and adequacy of indirect evidence linking intermediate or surrogate outcomes to our outcomes of interest.
Assessing the Relative Magnitude of Risks and Benefits
Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits. Health outcomes are one of several considerations in determining whether an item or service is reasonable and necessary. CMS places greater emphasis on health outcomes actually experienced by patients, such as quality of life, functional status, duration of disability, morbidity and mortality, and less emphasis on outcomes that patients do not directly experience, such as intermediate outcomes, surrogate outcomes, and laboratory or radiographic responses. The direction, magnitude, and consistency of the risks and benefits across studies are also important considerations. Based on the analysis of the strength of the evidence, CMS assesses the relative magnitude of an intervention or technology’s benefits and risk of harm to Medicare beneficiaries.


