Administrative File: CAG-00453N
FROM: Tamara Syrek Jensen, JD
Director, Coverage and Analysis Group
Joseph Chin, MD, MS
Deputy Director, Coverage and Analysis Group
Steven A. Farmer, MD, PhD
Chief Strategy Officer
Melissa Evans, PhD, MSAE
Director, Division of Policy and Evidence Review
Lori Ashby, MA
Deputy Director, Division of Policy and Evidence Review
David Dolan, MBA
Lead Analyst
Rachel Katonak, PhD, RN
Analyst
Patricia Bright, PhD, MSPH, RN
Lead Epidemiologist
Joseph Dolph Hutter, MD, MA
Lead Medical Officer
SUBJECT: National Coverage Determinations for Artificial Hearts and related devices (§20.9) and Left Ventricular Assist Devices (§ 20.9.1) (CAG-00453N)
DATE: December 1, 2020
I. Decision
A. The Centers for Medicare & Medicaid Services (CMS) is making changes to two separate, but medically related, National Coverage Determinations (NCDs). Given new evidence in the peer-reviewed medical literature, we are removing the NCD for Artificial Hearts and Related Devices established at §20.9 of the Medicare National Coverage Determination Manual. We are also revising the NCD at § 20.9.1 that provides coverage for Ventricular Assist Devices (VADs) for Bridge-to-Transplant and Destination Therapy. We summarize the changes below and fully explain the changes in the Analysis section of this NCD decision memo.
Artificial Hearts
CMS is removing the NCD at § 20.9, ending coverage with evidence development for artificial hearts and permitting Medicare coverage determinations for artificial hearts to be made by the Medicare Administrative Contractors (MACs) under § 1862(a)(1)(A) of the Social Security Act.
Ventricular Assist Devices
The decision is limited to durable, intracorporeal, left ventricular assist devices (LVADs), and does not include temporary VADs or extracorporeal membrane oxygen (ECMO). CMS is establishing coverage according to the following conditions:
Patient Selection Criteria
- Removes the current therapeutic intent-to-treat criteria of bridge-to-transplant (BTT) and destination therapy (DT), by removing the following BTT requirements:
- The requirement that the patient is active on the waitlist maintained by the Organ Procurement and Transplantation Network (OPTN).
- The requirement that the implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved transplant center prior to implantation of the VAD.
- Extends evidence based patient selection criteria, that previously applied only to DT, for all LVAD procedures for short-term (e.g., bridge-to-recovery and bridge-to-transplant) or long-term (e.g., destination therapy) mechanical circulatory support (see below in covered indications).
- Modifies patient selection criteria to reflect the major inclusion criteria of contemporary trials.
Facility criteria
- The facility criteria remain unchanged from the current NCD. The removal of therapeutic intent is not expected to decrease appropriate patient access to LVADs.
Below is the 20.9.1 NCD (Ventricular Assist Devices). All proposed changes are in italics. Any part of 20.9.1 that is not italicized is not proposed to be revised.
B. Covered Indications
1. Advanced Heart Failure
Left ventricular assist devices (LVADs) are covered if they are FDA approved for short-term (e.g., bridge-to-recovery and bridge-to-transplant) or long-term (e.g., destination therapy) mechanical circulatory support for heart failure patients who meet the following criteria:
- Have New York Heart Association (NYHA) Class IV heart failure; and
- Have a left ventricular ejection fraction (LVEF) ≤ 25%; and
- Are inotrope dependent
OR
have a Cardiac Index (CI) < 2.2 L/min/m2, while not on inotropes, and also meet one of the following:
- Are on optimal medical management (OMM), based on current heart failure practice guidelines for at least 45 out of the last 60 days and are failing to respond; or
- Have advanced heart failure for at least 14 days and are dependent on an intra‐aortic balloon pump (IABP) or similar temporary mechanical circulatory support for at least 7 days.
Beneficiaries receiving a VAD must be managed by an explicitly identified, cohesive, multidisciplinary team of medical professionals with appropriate qualifications, training, and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in informed decision making. The team members must be based at the facility and must include individuals with experience working with patients before and after placement of a VAD.
The team must include, at a minimum:
- At least one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 durable, intracorporeal, left ventricular assist devices over the course of the previous 36 months with activity in the last year.
- At least one cardiologist trained in advanced heart failure with clinical competence in medical- and device-based management including VADs, and clinical competence in the management of patients before and after placement of a VAD.
- A VAD program coordinator.
- A social worker.
- A palliative care specialist.
The process for organizations to apply for CMS approval to be designated as a credentialing organization for VAD facilities is posted on our web site along with a list of approved credentialing organizations, approved standard versions, and credentialed facilities: http://www.cms.gov/Medicare/Medicare-General-Information/MedicareApprovedFacilitie/VAD-Destination-Therapy-Facilities.html
See Appendix B for the manual language.
II. Background
Throughout this document we use numerous acronyms, some of which are not defined as they are presented in direct quotations. Please find below a list of these acronyms and corresponding full terminology:
6-MWD – six-minute walk distance
6-MWT – six-minute walk test
ACC – American College of Cardiology
ACCF – American College of Cardiology Foundation
ACE – Angiotensin-converting enzyme inhibitor
ACGME - Accreditation Council for Graduate Medical Education
ACP – American College of Physicians
AHA – American Heart Association
ARB – angiotensin receptor blocker
BiVAD – biventricular assist device
BMI – body mass index
BSA – body surface area
BTC – bridge to candidacy
BTT – bridge to transplant
CAP – continued access protocol
CDC – Centers for Disease Control and Prevention
CMS – Centers for Medicare & Medicaid Services
CRT – Cardiac resynchronization therapy
DNV – Det Norske Veritas Healthcare Inc.
DT – destination therapy
EQ-5D – EuroQuol-5D
EQ-5D VAS – EuroQol-5D Visual Analog Scale
FDA – Food and Drug Administration
HFrEF – heart failure with reduced ejection fraction
HFSA – Heart Failure Society of America
HM II – HeartMate II Left Ventricular Assist System
HM VE – HeartMate Vented Electric Left Ventricular Assist System
HM XVE – HeartMate XVE
HRQOL - health-related quality of life
HRSA – Health Resources and Services Administration
HW VAS – HeartWare Ventricular Assist System
IABP – intraaortic balloon pump
ICD – implantable cardioverter defibrillator
INTERMACS – Interagency Registry for Mechanically Assisted Circulatory Support
ISHLT - International Society for Heart and Lung Transplantation
LVAD – left ventricular assist device
LVEF – left ventricular ejection fraction
KCCQ – Kansas City Cardiomyopathy Questionnaire
KCCQ CSS – Kansas City Cardiomyopathy Questionnaire Clinical Summary Score
KCCQ OSS – Kansas City Cardiomyopathy Questionnaire Overall Summary Score
MAC - Medicare Administrative Contractor
MCS – mechanical circulatory support
MCSD – mechanical circulatory support device
MEDCAC – Medicare Evidence Development and Coverage Advisory Committee
METS – metabolic equivalent task score
MLHFQ – Minnesota Living with Heart Failure Questionnaire
NCA – National Coverage Analysis
NCD – National Coverage Determination
NHLBI – National Heart, Lung and Blood Institute
NIH – National Institutes of Health
NYHA – New York Heart Association
OPTN – Organ Procurement and Transplantation Network
PMA – premarket approval
PROs – patient-reported outcomes
QOL – quality of life
REMATCH – Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure
UNOS – United Network for Organ Sharing
VAD – ventricular assist device
Heart Failure
Heart failure is a condition in which the heart cannot pump blood adequately to meet the body’s needs at rest or with exertion. About 6.5 million people in the United States have heart failure which is associated with significant mortality, morbidity, and healthcare expenditures, particularly among those aged ≥ 65 years old (CDC, 2019; Roger, 2013). In addition to age, which is an independent risk factor for heart failure, older adults often have additional risk factors such as high blood pressure, diabetes mellitus, coronary heart disease, tobacco use, and overweight/obesity and may have been exposed to these risk factors for many years.
When the heart fails to adequately pump blood, excess fluid is retained and tissues do not get enough oxygen. This results in symptoms such as shortness of breath, swelling of the legs, and fatigue and causes substantial morbidity and, in the most serious circumstances, mortality. The leading cause of hospitalization among older adults is heart failure and Medicare beneficiaries with heart failure have the highest readmission rate of any condition (Jackson et al., 2018). Heart failure contributes to at least 287,000 deaths annually in the United States (Emory Healthcare, 2019).
While there are objective measures of the severity of heart failure such as ejection fraction and cardiopulmonary exercise testing, care is most often driven by symptom-based classifications including the New York Heart Association (NYHA) Functional Classification, INTERMACS profiles, and the American Heart Association and American College of Cardiology (AHA/ACC) Stages of Heart Failure.
NYHA Classification
The NYHA Functional Classification is a subjective measure of the severity of heart failure symptoms which some have criticized as being unresponsive to change, having a high degree of interobserver variability, and providing the perspective of the doctor rather than the patient (Green, Porter, & Bresnahan, 2000; Miller & Guglin, 2013). The four NYHA classes include:
Class I: Patients with cardiac disease but without resulting limitation of physical activity. Ordinary physical activity does not cause undue fatigue, palpitation, dyspnea or anginal pain.
Class II: Patients with cardiac disease resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity results in fatigue, palpitation, dyspnea or anginal pain.
Class III: Patients with cardiac disease resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes fatigue, palpitation, dyspnea or anginal pain.
Class IV: Patients with cardiac disease resulting in inability to carry on any physical activity without discomfort. Symptoms of heart failure or the anginal syndrome may be present even at rest. If any physical activity is undertaken, discomfort increases.
INTERMACS profiles were developed to further classify patients with advanced NYHA class III and class IV heart failure into one of seven profiles (Stevenson et al., 2009):
Profile 1 - Critical cardiogenic shock: Patient with life-threatening hypotension despite rapidly escalating inotropic support, critical organ hypoperfusion, often confirmed by worsening acidosis and/or lactate levels.
Profile 2 – Progressive decline: Patient with declining function despite intravenous inotropic support, may manifest with worsening renal function, nutritional depletion, or inability to restore volume balance. Also describes declining status in patients unable to tolerate inotropic therapy.
Profile 3 - Stable but inotrope dependent: Patient with stable blood pressure, organ function, nutrition, and symptoms on continuous intravenous inotropic support (or a temporary circulatory support
device or both), but demonstrating repeated failure to wean from support due to recurrent symptomatic hypotension or renal dysfunction.
Profile 4 - Resting symptoms: Patient who can be stabilized close to normal volume status but experiences daily symptoms of congestion at rest or during activities of daily living. Doses of diuretics generally fluctuate at very high levels.
Profile 5 - Exertion intolerant: Patient who is comfortable at rest and with activities of daily living but is unable to engage in any other activity, living predominantly within the house.
Profile 6 - Exertion limited: Patient without evidence of fluid overload who is comfortable at rest, and with activities of daily living and minor activities outside the home but fatigues after the first few minutes of any meaningful activity.
Profile 7 - Advanced NYHA class III: Patient who is clinically stable with a reasonable level of comfortable activity, usually able to walk more than a block. Has a history of previous decompensation but any decompensation requiring intravenous diuretics or hospitalization within the previous month should make this person a patient profile 6 or lower.
The AHA/ACC Stages emphasize the development and progression of heart failure ranging from Stage A (risk factors but no current cardiac abnormality) to Stage D (refractory heart failure, may be eligible for advanced treatments such as continuous inotropes, heart transplant, ventricular assist device placement (VAD), or end-of-life care) (Yancy et al., 2017).
Therapeutic interventions include modification of diet and lifestyle (such as restricting dietary sodium intake or increasing exercise) and medications including diuretics, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), aldosterone antagonists, beta-blockers, or digoxin. In the most severe cases, intravenous inotropic medications can be used. Inotropic medications do not reverse heart failure but may improve symptoms by making the heart beat stronger or reducing strain on the heart by reducing blood pressure. Device therapies include implantable cardioverter defibrillators (ICDs) for patients at risk for sudden cardiac death; pacemakers and cardiac resynchronization devices for patients with abnormalities in the heart’s electrical conduction system; artificial hearts, and LVADs which are the subject of this analysis.
Heart failure can be a progressive disease with increasing symptoms over time despite optimal medical management, though the time course is difficult to predict. Eventually, the heart fails completely and can no longer pump enough blood to sustain life. At this end-stage, eligible patients can undergo heart transplant; however, due to lack of available organs for transplant, many patients clinically decline or die on the transplant waitlist while awaiting life-saving transplantation (OPTN, 2019). In addition, older patients are often not eligible for heart transplant due to comorbid conditions, greatly increasing the risk of poor outcomes.
Mechanical Circulatory Support
There are a number of different mechanical devices that may be used to support the ventricles of a failing heart on either a temporary or a permanent basis. An artificial heart is an implanted prosthetic device that replaces the heart. Ventricular assist devices (VADs) or left ventricular assist devices (LVADs) are mechanical blood pumps that are surgically attached to one or both intact ventricles of a damaged or weakened native heart to assist in pumping blood. A relatively small proportion of patients suffer from biventricular failure. The artificial heart is a biventricular replacement device. Use of an artificial heart requires removal of both ventricles and removal of the device is not possible. In contrast, the heart remains intact with VADs with the possibility for the native heart to recover and for removal of the device.
Patients who may be candidates for LVAD implant undergo extensive clinical testing to ensure an adequate severity of heart failure but acceptable severity of comorbidities. This evaluation attempts to balance the benefits that might be achieved by LVAD implant with the significant risks of the surgery and prolonged device support. Initially, LVADs were used in the hospital as short-term support for patients with acute heart failure caused by temporary conditions such as infection or open heart surgery. With the development of smaller implantable pumps, patients could be ambulatory, discharged from the hospital, and supported on device for longer periods of time. These durable LVADs were first introduced in patients on the heart transplant waitlist as a “bridge to transplant (BTT)” since the duration of support was intended to be finite. With heart transplants in limited supply and additional clinical experience gained, devices were subsequently implanted as “destination therapy (DT)” in patients who are ineligible for heart transplant who required permanent support. Heart failure patients that are candidates for LVAD therapy who are not classified as either BTT or DT at the time of LVAD implant are referred to as bridge to decision or bridge to candidacy (BTC). Some patients transition between the categories of BTT, BTC, and DT over time with the development or resolution of comorbid conditions. In addition, this categorization can be inconsistent due to differences in transplant approval and listing processes at individual transplant hospitals.
The use of artificial hearts and related devices, including VADs, as a therapeutic tool has continued to evolve since our last national coverage analyses for BTT and DT. For VADs (NCD 20.9.1), the scope of this national coverage analysis (NCA) is limited to the coverage requirements for BTT and DT of durable LVADs for end-stage congestive heart failure. Specifically, we review the evidence for whether it supports changes to our current patient selection and facility criteria.
III. History of Medicare Coverage
Beginning in 1986, artificial hearts were non-covered under the Medicare program. In 2008, CMS began covering artificial hearts for bridge-to-transplant and destination therapy under coverage with evidence development for beneficiaries enrolled in a clinical study that meets all of the criteria specified in the NCD.
In 1996, CMS began covering VADs implanted as BTT at Medicare-approved heart transplant centers. In 2001, CMS covered implantation at sites other than Medicare-approved heart transplant centers. Currently, devices are covered only if they have received approval from the FDA for that purpose, and the VADs are used according to FDA-approved labeling instructions.
In 2003, CMS began covering VADs implanted as DT at Medicare-approved heart transplant centers meeting specific facility criteria including participation in a national, audited registry. In 2007, CMS allowed implantation at sites other than Medicare-approved heart transplant centers, named INTERMACS as the required registry, and required facilities to be credentialed by the Joint Commission based on standards dated February 2007. DT is for patients that require mechanical cardiac support. Currently, the VADs used for DT are covered only if they have received approval from the FDA for that purpose. The current patient selection criteria became effective November 9, 2010, and the current facility criteria became effective October 30, 2013.
Since there are existing NCDs for artificial hearts and VADs, this review is a reconsideration of these current policies. The current policies are codified in sections 20.9 and 20.9.1 of the Medicare National Coverage Determinations manual. These sections of the NCD manual have been included in Appendix C and D.
A. Current Request
CMS received a complete, formal request from Syncardia to reconsider the coverage with evidence development (CED) requirement for artificial hearts for bridge-to-transplant based on new scientific evidence generated since the previous decision in 2008.
Concurrently, CMS received a complete, formal request from Abbott Vascular asking that we reconsider the LVADs NCD to remove the therapeutic intent-to-treat criteria (e.g., bridge-to-transplant and destination therapy) for left ventricular assist device (LVAD) candidacy, and base LVAD coverage requirements on patient characteristics from new scientific evidence generated since the previous reconsideration of this NCD in 2013. The scope of this reconsideration is limited to the coverage requirements for LVADs for bridge-to-transplant and destination therapy. CMS is not reconsidering any other section of NCD 20.9.1. The formal request letters can be viewed via the tracking sheet for this NCA on the CMS website at https://www.cms.gov/medicare-coverage-database/details/nca-tracking-sheet.aspx?NCAId=298.
B. Benefit Category
Medicare is a defined benefit program. For an item or service to be covered by the Medicare program, it must fall within one of the statutorily defined benefit categories outlined in the Social Security Act.
Artificial hearts qualify as:
- Inpatient Hospital Services
- Prosthetic Devices
LVADs qualify as:
- Inpatient Hospital Services
Note: This may not be an exhaustive list of all applicable Medicare benefit categories for this item or service.
IV. Timeline of Recent Activities
| Date | Action |
|---|
| February 3, 2020 |
CMS opens an NCA for Initial 30-day public comment period begins. |
| March 4, 2020 |
First public comment period ends. CMS receives 35 comments. |
| August 12, 2020 |
Proposed Decision Memorandum posted. 30-day public comment period begins. |
| September 11, 2020 |
30-day public comment period ends. CMS receives 42 comments. |
V. Food and Drug Administration (FDA) Status
The FDA approved the SynCardia Systems, Inc., CardioWest temporary Total Artificial Heart (hereinafter called the TAH-t) in 2004, which is indicated for use as a bridge to transplantation in cardiac transplant-eligible candidates at risk of imminent death from biventricular failure. The CardioWest TAH-t System is intended for use inside the hospital.
The FDA has also approved several durable VADs for use as bridge-to-recovery (BTR), BTT, and/or DT in adults since 1992, some of which are pulsatile-flow pumps and are no longer in clinical use, such as the ABIOMED BVS 5000 Bi-Ventricular Support System (1992), Thoratec HeartMate IP (1994), Thoratec HeartMate VE (1998), and Thoratec HeartMate XVE (2001).
Thoratec HeartMate II Left Ventricular Assist System (HM II) was the first continuous-flow blood pump, which received FDA approval for BTT in 2008 and for DT in 2010. The second continuous-flow blood pump approved by the FDA was the HeartWare HVAD, which was approved for BTT in 2012 and for DT in 2017. The latest continuous-flow blood pump approved by the FDA was the Thoratec HeartMate 3 Left Ventricular Assist System, which was first approved for providing short-term hemodynamic support (e.g., BTR or BTT) in 2017 and then for providing long-term hemodynamic support (e.g., DT) in 2018. All three of these VADs are in clinical use presently.
VI. General Methodological Principles
When making national coverage determinations, CMS generally evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. The critical appraisal of the evidence enables us to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for beneficiaries. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary.
A detailed account of the methodological principles of study design that the Agency utilizes to assess the relevant literature on a therapeutic or diagnostic item or service for specific conditions can be found in Appendix A.
Public comments sometimes cite published clinical evidence and give CMS useful information. Public comments that give information on unpublished evidence such as the results of individual practitioners or patients are less rigorous and therefore less useful for making a coverage determination. Public comments that contain personal health information will be redacted or will not be made available to the public. CMS responds in detail to the public comments on a proposed national coverage determination when issuing the final national coverage determination.
VII. Evidence
A. Discussion of Artificial Hearts Evidence
1. Artificial Hearts Evidence Question
Q1: Is the evidence sufficient to end coverage with evidence development for artificial hearts?
Artificial hearts are commonly referred to as total artificial hearts (TAH) due to there being only one device that is currently available on the market, which is the TAH-t. Data from patients receiving TAH are limited due to the extremely low volume of patients receiving such devices, as exemplified by the studies below:
A peer-reviewed study conducted to inform CMS through coverage with evidence development (CED) identified 450 patients (87% men) who underwent a temporary TAH implantation as BTT or as BTD in the INTERMACS database between June 2006 and April 2017 (Arabia, 2018). Overall 3-,6-, and 12-month survival rates were 73%, 62%, and 53%, respectively.
A 2016 study identified patients from the United Network of Organ Sharing database (a private, non-profit organization that manages U.S. organ transplants). They assessed patients aged >18 years’ old who underwent heart transplantation between January 2005 and December 2014 comparing them to patients receiving biventricular assist device support as BTT (Cheng, 2016). The investigators identified 212 patients who received TAH with a mean age of 50 years (87% men). The 30-day, one-, and three-year post-transplantation survival was 88%, 78%, and 67%, respectively, for patients with TAH support.
Investigators at the Division of Cardiology, Pauley Heart Center, Virginia Commonwealth University, studied 66 consecutive patients implanted with the TAH from 2006 through 2012 to assess outcomes by blinded, retrospectively adjudicated INTERMACS profiles (Shah, 2016). Survival after TAH implantation at 6 and 12 months was 76% and 71%, respectively.
CMS did not request an external technology assessment (TA) and a MEDCAC meeting was not convened on this issue.
B. LVADs Introduction
The evidence summarized in this section includes the peer-reviewed, published clinical research pertinent to the use of durable, implantable left ventricular assist devices for advanced heart failure, whether for short-term use in patients who end up receiving an orthotopic heart transplant, or for long-term use in patients who never receive a transplant. Our assessment focuses exclusively on the two key questions below.
1. LVAD Evidence Question(s)
Q1: Is the evidence sufficient to conclude that modifying the current patient selection criteria for LVAD implantation will improve health outcomes for Medicare beneficiaries?
Q2: Is the evidence sufficient to conclude that modifying the current facility criteria for LVAD implantation will improve health outcomes for Medicare beneficiaries?
2. External Technology Assessments
CMS did not request an external technology assessment (TA) on this issue.
3. Internal Technology Assessment
Literature Search Methods
CMS examined whether the evidence is adequate to support changes to our current patient selection or facility criteria for VADs. We included studies with publication dates on or after 2013 – February 2020. We searched the PubMed database and Cochrane library using the terms heart-assist device, heart assist pump, left ventricular assist device, LVAD, mechanical circulatory support, or INTERMACS; and bridge to transplant, bridge to candidacy, destination therapy, myocardial recovery, or heart transplantation. Additionally, we searched for guidelines and consensus statements relevant to our evidence questions. We limited our search to English language publications in humans over the age of 18. This search resulted in 985 publications, which we then reviewed by title and abstract to select papers potentially relevant to our evidence questions.
We excluded studies of devices not currently approved by the FDA, devices that are no longer marketed, pre-pivotal studies of approved devices, studies of fewer than 50 VAD recipients, retrospective single-center case series, studies reporting only intermediate or surrogate outcomes, studies only reporting outcomes following heart transplant, or studies of cost or cost effectiveness. We also excluded studies focused only on pulsatile, temporary (non-durable), percutaneous, right-sided, biventricular, or partial-support devices and studies of artificial hearts.
The clinical trials and observational studies that were relevant to our key questions and informed our decision language appear in the tables below.
Table 1. Key LVAD Trials and Registry Studies – Advanced Heart Failure
(Since the CMS 2013 NCD, with original reference trials in italics)
| Study |
Design |
P I C O |
Results |
| Year |
Study/Author |
Type |
Sites+ |
Size (N) |
Patients included |
Age
(yrs)
I;C |
Male
(%)
I;C |
Intervention |
Control |
Outcome
(Primary) |
Based on Primary Outcome |
| 2001 |
REMATCH
(Rose) |
RCT (1:1) |
20 US |
129 |
- DT (destination therapy; ineligible for heart transplant)
- NYHA class IV ≥ 90 days on OMT
- LVEF ≤ 25%
- Peak O2 consumption ≤12 ml per/kg/min; OR Inotrope dependent, decreasing renal function, or worsening pulmonary congestion
|
66; 68 |
78; 82 |
HeartMate 1 (pulsatile-flow) |
Medical Therapy |
Survival at 1 yr (overall) |
HM1 superior to medical therapy
HM1 52%
Medical 25%
(P = 0.002) |
| 2009 |
Heart Mate II
(Slaughter) |
RCT (2:1) |
38 US |
200 |
ΔT |
62; 63 |
81; 92 |
HeartMate II (axial-flow) |
HeartMate 1 (pulsatile-flow) |
Survival at 2 yrs free of disabling stroke and reoperation |
HMII superior to HM1
HMII 46%
HM1 11%
(P < 0.001) |
| 2014 |
HeartMate II DT FDA Post-Approval
(Jorde) |
Obser. |
61 US Regis;
34 US RCT |
380 |
- DT
- 1st 247 HMII DT patients in INTERMACS Registry v. 133 HMII DT patients in 2009 HMII RCT
|
70; 62 (≥60 yrs) |
83; 80 |
Community implant of HeartMate II (Registry) |
Clinical trial implant of HeartMate II (RCT) |
Survival at 2 yrs free of disabling stroke and reoperation |
HMII in community superior or similar to HMII in trial
Registry HMII 54%
RCT HMII 44%
(P = 0.042) |
| 2015 |
INTERMACS Annual Rpt-7th
(Kirklin) |
Obser. |
158 US |
15,745 |
- All devices
- Continuous-flow (CF): N=12,030
- Intent (CF devices): 27% BTT (bridge to transplant; awaiting heart transplantation) listed, 38% T
|
N/R |
N/R |
N/A |
N/A |
Survival at various yrs |
Survival (full cohort): 1 yr 80%; 2 yrs 70% Survival (DT, 2012-14): 1 yr 76%; 3 yrs 57% |
| 2017 |
ENDURANCE
(Rogers) |
RCT (2:1) |
48 US |
446 |
- DT
- NYHA class IIIB* or IV
- LVEF ≤ 25%
- On OMM, including dietary salt restriction and diuretics, 45 of the last 60 days and failing to respond; OR
- Advanced HF for at least 14 days, and dependent on IABP for 7 days and/or inotropes for at least 14 days
|
63.9; 66.2 |
76.4; 82.4 |
HeartWare (centrifugal-flow) |
HeartMate II (axial-flow) |
Survival at 2 yrs free of disabling stroke and reoperation~ |
HW centrifugal noninferior
HW 55.0%
HMII 57.4%
(P = 0.67) - HW with more strokes
- HMII with more reoperations
|
| 2018 |
Euromacs 2nd Rpt
(de By) |
Obser. |
52 Eur |
2681 |
- All devices
- Continuous-flow (CF), BTT or DT: N=2,113
|
51.7 |
82 |
N/A |
N/A |
Survival at various yrs |
- Survival (CF LVAD, BTT or DT):
1 yr 69%
2 yrs 55%
3 yrs 44% - BTT survival > DT
|
| 2019 |
MOMENTUM 3
(Mehra) |
RCT
(1:1) |
69 US |
1028 |
- DT, BTT, BTC (bridge to candidacy for transplant; not current transplant candidates but may become candidates for heart transplantation)
- NYHA class III with dyspnea upon mild physical activity, or NYHA class IV
- LVEF ≤ 25%
- Inotrope dependent; OR
Not inotrope dependent, but CI <2.2 L/min/m2, and either:
- on OMM 45 of last 60 days and failing to respond; OR
- advanced HF for at least 14 days and dependent on IABP for at least 7 days
|
59; 60 |
79.7; 81.8 |
HeartMate 3 (centrifugal) |
HeartMate II (axial) |
Survival at 2 yrs free of disabling stroke and reoperation |
HM3 centrifugal superior to HMII axial (driven by reoperations) 2-yr results:
HM3 76.9%
HMII 64.8%
(P < 0.001) 1-yr results:
HM3 84%
HMII 74.8%*
HM3 with lower adverse event rates for stroke, bleeding, and pump thrombosis |
| 2019 |
INTERMACS 2018 Annual Rpt
(Kormos) |
Obser. |
Natl US |
18,539
(2012-2017: 14,195) |
Isolated continuous-flow LVAD implanted (74% of total registry patients) Intent: 26% BTT, 43% DTLVAD: 78% axial, 22% centrifugal (1% HM3) |
57.1 |
78.8 |
N/A |
N/A |
Survival at yrs 1 and 5 |
Results for 2012-2017 era:
Survival (full cohort): 1 yr 83%; 5 yrs 46%Survival at 1 yr:
Centrifugal 85%
Axial 84%Survival at 5 yrs:
Centrifugal 49%
Axial 46% Survival (DT): 1 yr 80%; 3 yrs 59%
Results from 2008-2017:
Adverse events at 1 yr:
Centrifugal (1% HM3) with more neuro events and pump-related infections. Axial with more GI bleeds |
| 2020 |
MOMENTUM 3 Subgroup Analysis
(Goldstein) |
RCT Sub-group |
69 US |
1028
(1:1) |
Secondary analysis of M3 trial: outcomes of LVAD use by BTT/BTC or DT therapy intent NYHA class III with dyspnea upon mild physical activity, or NYHA class IVLVEF ≤ 25%Inotropes as in M3 trial above |
59; 60 |
79.7; 81.8 |
BTT/BTC patients (HM3 and HMII) |
DT patients (HM3 and HMII) |
Survival at 2 yrs free of disabling stroke and reoperation |
Superior effect on outcomes of HM3 over HMII was similar in BTT/BTC and T groups |
| 2020 |
INTERMACS 2019 Annual Rpt
(Teuteberg) |
Obser. |
Natl US |
13,016
(2014-2018) |
Isolated continuous flow LVADs: 53% axial, 37% centrifugal, 10% HM3
Intent:52% DT, 46% BTT 'Control' = Baseline era 2014-16 ‘Intervention’ = Contemporary era 2017-2018 |
57.1; 57 |
77.3; 78.6 |
2017-18 data |
2014-2016 data |
Survival at various yrs. |
Results for full cohort (2014-2018): Survival at 1 yr: 82%
Survival at 5 yrs: 47%
Overall survival and freedom from major adverse events higher with contemporary, centrifugal-flow full magnetic levitation devices (e.g., HM3). |
PICO is Population, Intervention, Control, Outcomes.
I = intervention, C = Control.
OMT = optimal medical therapy
RCT = randomized controlled trial
+ All trials and studies are multicenter.
^ Disabling stroke is Rankin scale >3 for all trials and studies.
~ Reoperation to remove or replace because of a failure of the original device
Table 2. LVAD Studies on Facility/Volume Requirements
(Since the CMS 2013 NCD)
| Study |
Design |
P I C O |
Results |
| Year |
Study / Author |
Type |
Sites+ |
Size (N) |
Patients included |
Age
(yrs) |
Male
(%) |
Intervention |
Control |
Outcome
(Primary) |
Based on Primary Outcome |
| 2016 |
Academic Centers: Outcomes and Volume
(Davis, Hohmann, Doukky, Levine, & Johnson) |
Obser. |
88 US |
7714 patients and 581 surgeons |
Academic Med Centers: any LVAD implant, DT or BTTSurvived v died by procedure volume |
Surv;Died
56; 59 |
Surv;Died 78%; 75 |
N/A |
N/A |
In-hospital all-cause mortality |
Surgeons’ LVAD procedure volume, not annual hospital procedure volume, are assoc. with in-hospital mortality |
| 2016 |
NIS: Outcomes and Volume
(Shah) |
Obser. |
Natl US |
1749 |
National Inpatient Sample: all LVAD outcomes by hospital volume tertiles |
55.4 |
77 |
N/A |
N/A |
In-hospital all-cause mortality; length of stay (LOS) |
High annual LVAD volume assoc. with lower in-hospital mortality and LOS; optimal volume threshold: >20 LVADs/yr |
| 2017 |
INTERMACS: Outcomes and Volume
(Cowger) |
Obser. |
Natl US |
2012-2014: 7416 |
INTERMACS registry (2012-2014): all LVAD outcomes by hospital volume quartiles |
>60 (49%) |
79 |
N/A |
N/A |
1 yr survival (overall) |
Results 2012-2014:
Very low annual LVAD volume (<10/yr), and high volume (>50/yr) assoc. with lower survival, compared to midrange hospitals (31-50/yr) |
| 2018 |
INTERMACS: Transplant Centers Better?
(Brinkley) |
Obser. |
Natl US |
3583 |
INTERMACS registry: outcomes at transplant centers (3323 patients) v non-transplant centers (260 patients)Isolated continuous-flow LVAD implanted for DT |
Cntr: Tr;NoTr
65; 65 |
Cntr: Tr;nonTr
81.3; 79.6 |
Non-transplant center |
Transplant center |
“Survival, censored at device explant for recovery or transplant” |
DT outcomes similar at transplant and non-transplant centers for: 30 day and 1 yr survival; death or major adverse events.
Patients at transplant centers were sicker |
4. Medicare Evidence Development & Coverage Advisory Committee (MEDCAC)
A MEDCAC meeting was not convened on this issue.
5. Evidence-Based Guidelines
The pertinent evidence-based guidelines are summarized below (in order of most recent).
Kirklin JK, Pagani FD, Goldstein DJ, et al. American Association for Thoracic Surgery/International Society for Heart and Lung Transplantation guidelines on selected topics in mechanical circulatory support (Guidelines) J Heart Lung Transplant. 2020 Mar; 39(3):187-219.
This is the most recently published professional society guideline. The guideline covers a range of topics from preoperative evaluation through surgical approach and management and draws on multiple sources including results of the two interim reports of the MOMENTUM 3 trial.
The guideline states that the, “Preoperative evaluation of a patient considered for implantation of a durable LVAD begins with assessment of the criteria that define the indication for implant (e.g., bridge to transplant [BTT], bridge to candidacy, or destination therapy [DT]).”
As a general conclusion in the section on preoperative evaluation and optimization, the authors state, “Although individual organ system dysfunction may be an absolute contraindication for VAD surgery, a decision to implant will often involve a cumulative assessment of several relative contraindications and the associated overall risk.”
The guideline notes that in the setting of a maximal study (respiratory exchange ratio >1.1), peak oxygen consumption <14 mL/kg/min (or <50% of predicted, whichever is lowest), and/or a ventilation/carbon dioxide production slope >36 have been associated with marked impairment in cardiac reserve and poor short- to intermediate-term prognosis. Most US payers use these values as qualifying criteria for destination therapy LVAD implantation.
Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. 28 Apr 2017 https://doi.org/10.1161/CIR.0000000000000509 Circulation. 2017;136:e137–e161.
The 2013 guidelines, to which this is a 2017 focused update, was previously reviewed in our 2013 NCD. The update briefly addresses LVADs or MCS. Symptomatic patients with refractory NYHA class III-IV (stage D) should be considered for LVAD, transplant, or palliative care:
FIGURE 1 Treatment of HFrEF Stage C and D (Yancy et al., 2017)
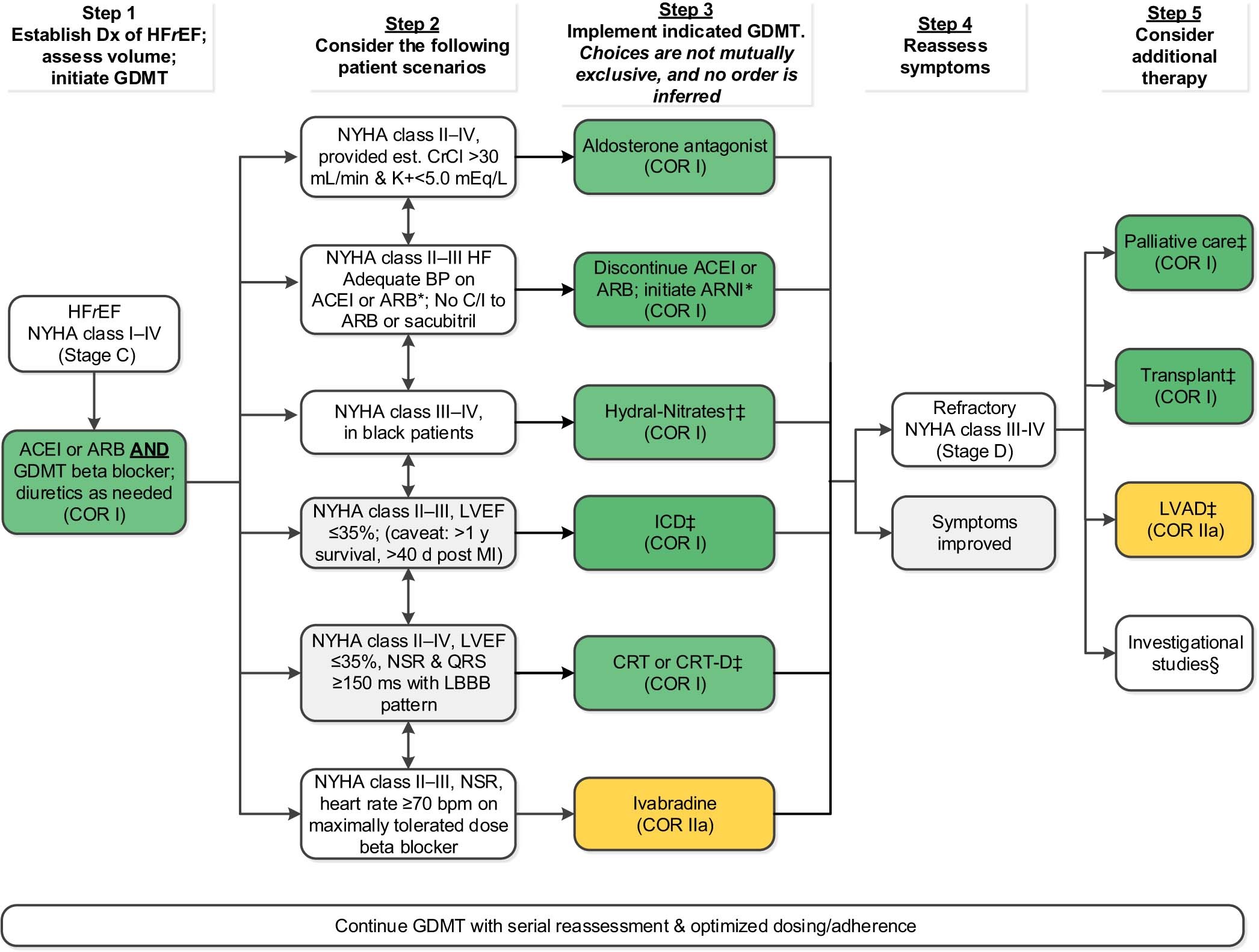
For all medical therapies, dosing should be optimized and serial assessment exercised.
*See text for important treatment directions.
†Hydral-Nitrates green box: The combination of ISDN/HYD with ARNI has not been robustly tested. BP response should be carefully monitored. ‡See 2013 HF guideline(9).
§Participation in investigational studies is also appropriate for stage C, NYHA class II and III
HF.
ACEI indicates angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor-blocker; ARNI, angiotensin receptor-neprilysin inhibitor; BP, blood pressure; bpm, beats per minute; C/I, contraindication; COR, Class of Recommendation; CrCl, creatinine clearance; CRT-D, cardiac resynchronization therapy–device; Dx, diagnosis; GDMT, guideline-directed management and therapy; HF, heart failure; HFrEF, heart failure with reduced ejection fraction; ICD, implantable cardioverter-defibrillator; ISDN/HYD, isosorbide dinitrate hydral-nitrates; Kþ, potassium; LBBB, left bundle-branch block; LVAD, left ventricular assistdevice; LVEF, left ventricular ejection fraction; MI, myocardial infarction; NSR, normal sinus rhythm; and NYHA, New York Heart Association.
In a companion slide presentation of this focused update, the authors describe “Stage D Refractory [heart failure]” as “e.g., patients with: Marked [heart failure] symptoms at rest; Recurrent hospitalizations despite GDMT” [guideline directed management and therapy].
NICE. Implantation of a left ventricular assist device for destination therapy in people ineligible for heart transplantation. Interventional procedures guidance. Published: 27 March 2015 www.nice.org.uk/guidance/ipg516.
The NICE guidance is general, and emphasizes team care:
1 Recommendations
1.1 Current evidence on the efficacy and safety of the implantation of a left ventricular assist device for destination therapy in people ineligible for heart transplantation is adequate to support the use of this procedure provided that normal arrangements are in place for clinical governance, consent and audit.
1.2 Patient selection should be done by a multidisciplinary team that includes a cardiologist with a specialist interest in heart failure, a cardiothoracic surgeon and a cardiac anesthetist (see section 1.3).
1.3 Implantation of left ventricular assist devices for destination therapy should be done by surgeons, anesthetists and intensive care specialists with special training and regular practice in performing this procedure and caring for these patients. Subsequent care should be provided by a multidisciplinary team including staff with the expertise to deal with patients' medical and psychological management, and with the maintenance of their left ventricular assist devices.
6.1 The Committee noted that heart failure is very common. It considered that the use of left ventricular assist devices for destination therapy in people ineligible for heart transplantation needs very careful selection of patients who are likely to derive sustained benefit in terms of survival and quality of life.
6.2 The Committee recognized that this procedure is associated with a high incidence of complications, but it judged that the potential benefit for appropriately selected patients outweighed its potential for harm.
6.3 The Committee noted that technology for this procedure has evolved significantly in recent years and continues to do so.
Note that the corresponding NICE guidance from 2006 for LVAD use for
short-term circulatory support, at https://www.nice.org.uk/guidance/ipg177/chapter/2-The-procedure is very similar, also emphasizes team care, and adds these comments specific to short-term use:
2.5.1 This guidance refers to patients for whom other treatments such as intra-aortic balloon pumping would be ineffective, who are considered eligible for heart transplantation, or who have acute severe heart failure that is likely to be reversible (such as acute myocarditis). It was noted that other patients might potentially benefit from this procedure, such as patients who cannot be weaned off cardiopulmonary bypass after cardiac surgery and patients with cardiogenic shock after acute myocardial infarction.
6. Professional Society Recommendations / Consensus Statements / Other Expert Opinion
(in order of most recent)
Potapov EV, Antonides C, Crespo-Leiro MG, et al. 2019 EACTS Expert Consensus on long-term mechanical circulatory support. Eur J Cardiothorac Surg. 2019 Aug 1;56(2):230-270. doi: 10.1093/ejcts/ezz098.
The European Association for Cardio-Thoracic Surgery (EACTS) state, “The evidence published so far is insufficient to allow for generation of meaningful guidelines complying with EACTS requirements. Instead, the EACTS presents an expert opinion in the LT-MCS field.”
The authors provided the following background: “Long-term durable mechanical circulatory support (LT-MCS) has evolved significantly in the last decade. Today’s devices have become more reliable, and their durability has increased whereas device related complications have drastically decreased compared with earlier generations of devices.”
They also support a change in terminology: “In this statement, we have generally refrained from using the designations of bridge to transplant and destination therapy in accordance with the more recent randomized trials in this field.”
A central purpose of this expert consensus is to provide patient “selection criteria for evaluation of potential candidates.” As a summary of past (not the most recent) trials, they state that “Major trials have established the efficacy of LVADs in patients with a low left ventricular ejection fraction (<25%), who were inotrope dependent or were persistently New York Heart Association (NYHA) functional class IIIb or IV despite optimal medical therapy. Additionally, a maximal oxygen consumption below 12 ml/kg/min was often used as an inclusion criterion.”
The table below summarizes their current recommendations on specific patient characteristics.
Table 3. EACTS Expert Consensus on long-term mechanical circulatory support
| Recommendation | Class | Level | References |
|---|
| It is recommended that reversible causes of heart failure are ruled out. |
I |
B |
|
LT-MCS implantation should be considered in patients with the following:
- New York Heart Association functional class IIIB–IV and
- Ejection fraction ≤ 25% and
At least one of the following criteria:
- INTERMACS 2–4
- Inotrope dependence
- Progressive end-organ dysfunction
- Peak VO2 <12 ml/kg/min
- Temporary MCS dependence
|
IIa |
B |
|
LT-MCS implantation should be considered in patients with the following:
- New York Heart Association functional class IIIB–IV and
- Ejection fraction ≤ 25% and
At least one of the following criteria:
- INTERMACS 2–4
- Inotrope dependence
- Progressive end-organ dysfunction
- Peak VO2 <12 ml/kg/min
- Temporary MCS dependence
|
IIa |
B |
|
Patient characteristics associated with a high risk of poor outcome post-left ventricular assist device |
LT-MCS in patients with advanced age, after careful evaluation of comorbidities and frailty, should be considered. |
IIa |
C |
[3, 22-25] |
LT-MCS in patients with peripheral vascular disease, depending on its severity, may be considered. |
IIb |
C |
|
LT-MCS in patients with active systemic bacterial/fungal infection is not recommended. |
III |
B |
[26,27] |
In patients with well controlled HIV, hepatitis B or hepatitis C, LT-MCS should be considered. |
IIa |
B |
[26,27] |
In patients with diabetes with poor glycaemic control or end-organ complications, LT-MCS may still be considered. |
IIb |
B |
[22, 28-30] |
LT-MCS may be considered in patients with chronic dialysis. |
IIb |
C |
[31-34] |
LT-MCS implantation in patients with haemostatic deficiencies and coagulopathies may be considered. |
IIb |
B |
[35-38] |
LT-MCS implantation in patients with untreated aortic regurgitation or mechanical aortic valve is not recommended. |
III |
C |
[39,40] |
LT-MCS in patients with untreated severe mitral stenosis is not recommended. |
III |
C |
|
LT-MCS implantation in patients with irreversible liver dysfunction, as diagnosed by liver enzyme laboratory tests and the Model of End-stage Liver Disease score, is generally not recommended. |
III |
B |
[41] |
In patients with poor neurological and cognitive function, LT-MCS implantation is not recommended. |
III |
B |
[42,43] |
Frail patients and patients with limited mobility may, after careful evaluation, be considered for LT-MCS implantation. |
IIb |
B |
[44-48] |
LT-MCS in patients who are living alone or who are suffering from depression should, after careful evaluation, be considered. |
IIa |
C |
[19, 49-53] |
LT-MCS implantation in patients who suffer from dementia is not recommended. |
III |
C |
[19, 49-53] |
LT-MCS implantation in patients with active substance abuse, not willing to cease the abuse, is not recommended. |
III |
C |
|
LT-MCS implantation in patients with malignancies may be considered if expected survival is > 1 year. |
III |
C |
|
HIV: human immunodeficiency virus; INTERMACS: Interagency Registry for Mechanically Assisted Circulatory Support; LT-MCS: long-term mechanical circulatory support.
Cook JL, Colvin M, Francis GS, et al. Recommendations for the Use of Mechanical Circulatory Support: Ambulatory and Community Patient Care: A Scientific Statement From the American Heart Association. Circulation. 2017 Jun 20;135(25):e1145-e1158. doi: 10.1161/CIR.0000000000000507. Epub 2017 May 30.
Intended as recommendations to frontline community practitioners and first responders, the table on “Indications and Contraindications to Durable Mechanical Support” is relevant to this NCD. The criteria derive from past clinical guidelines and regulatory requirements (including the CMS 2013 NCD).
Table 4. Indications and Contraindications to Durable Mechanical Support
Indications: combination of the following:
Frequent hospitalizations for heart failure
NYHA class IIIb–IV functional limitations despite maximal therapy
Intolerance of neurohormonal antagonists
Increasing diuretic requirement
Symptomatic despite CRT
Inotrope dependence
Low peak VO2 (<14–16)
End-organ dysfunction attributable to low cardiac output
Contraindications
Absolute
Irreversible hepatic disease
Irreversible renal disease
Irreversible neurological disease
Medical non-adherence
Severe psychosocial limitations
Relative
Age >80 y for DT
Obesity or malnutrition
Musculoskeletal disease that impairs rehabilitation
Active systemic infection or prolonged intubation
Untreated malignancy
Severe PVD
Active substance abuse
Impaired cognitive function
Unmanaged psychiatric disorder
Lack of social support
____________________________________________________________
CRT indicates cardiac resynchronization therapy; DT, destination
therapy; NYHA, New York Heart Association; VO2, oxygen consumption; and
PVD, peripheral vascular disease.
7. Public Comment
Public comments sometimes cite the published clinical evidence and give CMS useful information. Public comments that give information on unpublished evidence such as the results of individual practitioners or patients are less rigorous and therefore less useful for making a coverage determination. CMS uses the initial public comments to inform its proposed decision. CMS responds in detail to the public comments on a proposed decision when issuing the final decision memorandum. All comments that were submitted without personal health information may be viewed in their entirety by using the following link: https://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=298.
Initial Comment Period: 02/03/2020 – 03/04/2020
During the initial 30-day public comment period, CMS received 35 comments. Of these 35 comments, one was omitted from publication on the CMS website due to a commenter posting a duplicative comment.
Artificial Hearts
Five commenters offered suggested changes to the 2013 NCD language. The commenters asked that CMS update the nationally covered indications based on newly available evidence and remove the coverage with evidence development criteria.
The majority of comments were provided by physicians/cardiologists and other healthcare professionals. One comment was from the AHA and one from the manufacturer of artificial hearts, Syncardia.
VADs
Thirty comments offered suggested changes to the NCD language for VADs. The majority of these comments asked for CMS to update the nationally covered indications based on newly available evidence from RCTs, with a number of commenters specifically referencing the MOMENTUM 3 study. The commenters asked that CMS remove the therapeutic intent-to-treat to criteria (bridge-to-transplant and destination therapy) and to base coverage on patient characteristics.
The majority of comments were provided by physicians and other healthcare professionals. There were two comments that represented five professional societies, including the American Heart Association (AHA), Society of Thoracic Surgeons, American College of Cardiology, American Association for Thoracic Surgery, and Heart Failure Society of America. Additional groups who offered comments were the University of Kansas Mechanical Circulatory Support program, Maine Medical Center, and MedStar Heart and Vascular Institute. We also received four comments from industry, including Medtronic, Abbott, AdvaMed, and DNV GL Healthcare.
Second Comment Period: 08/07/2020 – 09/11/2020
During the 30-day comment period following the release of the proposed decision memorandum, CMS received 42 comments. Eleven of the public comments referred to both artificial hearts and VADs. For artificial hearts, the majority of the comments supported removal of the current CED requirement, with one comment supporting coverage of artificial hearts at the discretion of the MACs. Most of the comments supporting the removal of the CED requirement suggested that CMS should continue to have a
national coverage determination for artificial hearts. For VADs, the majority of comments expressed support for the proposed decision to remove the current therapeutic intent-to-treat criteria of bridge-to-transplant (BTT) and destination therapy (DT), with some comments suggesting modifications to the proposed patient criteria.
The comments included 23 from physicians and other healthcare professionals; three from the public/non-affiliated individuals; and nine from medical facilities. Three comments represented seven national associations/professional societies, including the American Heart Association, American Stroke Association, Society of Thoracic Surgeons, American College of Cardiology, Heart Failure Society of America, American Association for Thoracic Surgery, and American Association of Heart Failure Nurses. We also received four comments from industry, including SynCardia Systems LLC, Medtronic, Abbott, and AdvaMed.
Comments Citing New Papers
Commenters cited 15 publications of studies relevant to this NCD that we had not reviewed at the time of publication of our proposed decision memorandum. One publication was the most recent annual report of the Intermacs registry (Teuteberg 2020); we believe this generally supports our current decision, and it has been added to Table 1 on “Key LVAD Trials and Registry Studies – Advanced Heart Failure,” and referenced it in our Analysis section. Several papers supported shared decisionmaking as well as the interrelated topics of the LVAD volume/outcomes association. These comments have been summarized and addressed below.
Artificial hearts
Medicare Administrative Contractor (MAC) Discretion
Comment: Many commenters expressed concerns that removing the NCD for artificial hearts would create inconsistent coverage, and could delay or limit access of a time-sensitive procedure for Medicare beneficiaries. Some commenters also stated that hospitals would be taking a financial risk by implanting patients with an artificial heart prior to authorization, and some hospitals may refuse to implant. Some commenters also stated that artificial hearts should not be precluded from a national coverage determination for the fact that it is a low volume procedure.
Response: We understand and appreciate the concerns regarding access to TAH. We believe this decision allows for appropriate coverage of TAH, ending the requirement under coverage with evidence development (CED) in which Medicare only covered TAH for patients enrolled in a CMS approved study. In general, we agree that no specific volume percentage should be a hard threshold that automatically triggers either MAC discretion or an NCD. However, extremely low volume directly limits the evidence base. After reviewing the evidence that was provided in public comments that supported national coverage of TAH, the available evidence does not support establishment of a clear set of criteria for national coverage unlike the case with LVADs building on MOMENTUM 3 and other trials. Given the small study samples, a Medicare patient who could benefit from TAH may be potentially excluded by a national coverage determination, but could receive one through a more individualized MAC assessment.
Additionally, we note that MACs have a history of handling coverage for acute scenarios at the local level. We do not believe that coverage of TAH will be delayed or limited. Some contractors may also choose to develop local coverage determinations (LCDs).
The MACs are structured to be able to take into account a beneficiary’s particular clinical circumstances which are especially important when overall prevalence is very low. As such, CMS continues to believe coverage of artificial hearts is an appropriate determination made by the MACs and will lead to greater access to artificial hearts under this expansion of coverage.
Comment: One commenter stated that the field of biventricular support is in its infancy and the proposed decision risks hindering important advances in biventricular support research. Other commenters believe CED specifically is needed to promote research with a greater focus on patient outcomes.
Response: While we encourage the full breadth of research suggested by commenters, CMS believes this could be better supported and guided by other agencies, and through a systematic, comprehensive approach to all of heart failure research. As stated in our previous response, we believe that allowing the MACs to make the §1862(a)(1)(A) reasonable and necessary determination for artificial hearts is appropriate.
Bridge-to-Transplant (BTT)
Comment: One commenter stated that the BTT/DT distinction for artificial hearts has become clinically irrelevant, as CMS has recognized to be the case for LVADs in the proposed decision memo, and the BTT/DT distinction artificially limits access to a subset of the patient population experiencing biventricular failure.
Response: We appreciate the comment. For the reasons previously stated, coverage of artificial hearts will be determined by Medicare Administrative Contractors (MACs). We would like to note that the FDA has only approved the artificial heart for use as DT in adult patients.
VADs
Patient Criteria
Comment: Multiple commenters recommended alternative patient selection criteria. For example, one commenter recommended “using the UNOS criteria of PCWP >15 mmHg and simultaneous cardiac index < 2.2 L/min/m2 while on inotropic or temporary mechanical circulatory support OR < 1.8 L/min/m2 off therapy.” Another commenter stated that IABP dependency for 7 days is too long and suggested a timeframe of 3-5 days, depending on patient clinical status.
Response: We appreciate the thoughtful comments, but we are not modifying the final decision to incorporate these changes. Multiple patient and disease variables were evaluated by the trialists who selected the specific inclusion and exclusion criteria for the large, contemporary LVAD trials such as MOMENTUM 3. The results from those trials offer the strongest form of evidence; and patients most like those in the trials are the ones most likely to benefit from the intervention. It is unclear if patients meeting other criteria would or would not benefit. We believe our final decision reflects the most recent evidence-based criteria, and is most likely to improve health outcomes for Medicare beneficiaries.
Comment: One commenter requested clarification of our exact patient criteria for an LVAD, and specifically what is an “and” versus an “or” clause in the criteria.
Response: We appreciate this comment and added clarifying “and/or” language to the patient criteria in section I of this final decision memorandum under “Covered Indications” for patients with
advanced heart failure.
Facility Criteria
Comment: Some commenters did not support CMS’ proposal to remove the requirement that the implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved transplant center prior to implantation of the VAD. They believe the removal of this requirement could inadvertently create a situation in which patients who could receive a heart transplant receive a LVAD instead, because they never got referred to a transplant physician and team for assessment, and thus never had the benefit of true, comprehensive patient-physician shared decision making (SDM). Other commenters provide further support for shared decision making for LVAD implantation.
Response: We recognize that heart transplant remains the gold standard treatment for severe, refractory heart failure. Currently, the primary care physician and/or general cardiologist for a patient with severe heart failure should consult a heart transplant physician for assessment. This NCD is specific to LVADs. The criteria for triggering that referral, the referral process itself, the evaluation of heart transplant candidates, organ donation waitlist criteria and other related processes, are outside the scope of this NCD. The focus of this NCD was to update patient selection criteria to reflect the current evidence, and as a result, to eliminate antiquated patient categories based on therapeutic intent at the time of LVAD implantation.
As for shared decision making, we strongly encourage the use of shared decision making generally, wherever it can be successfully applied, and have required shared decision making in other NCDs (for example, implantable cardiac defibrillators: https://www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=110&ncdver=4&NCAId=39). As part of SDM in current NCDs, we prefer to require the use of an evidence-based decision aid or tool. We support patient shared decision making in VADs, but there is not a fully developed tool available at this time. We strongly encourage standardized decision aids or tools to facilitate the decision making process between a patient and physician and will be monitoring this space closely.
Comment: One commenter stated that a more stringent requirement for VAD center volume is needed and that there are no data to support a specific surgeon volume.
Response: As we discuss in the analysis section, there has not been sufficient evidence published since the last reconsideration of this policy in 2013 to support a change to the facility requirements. A number of studies have provided data on volume, yet this newer evidence as a whole has been inconclusive. Several studies evaluated LVAD volume as a predictor of mortality, but did not standardize these volume assessments. Some evaluated surgeon volume as distinct from hospital volume. Others used hospital volume as a broad measure that might encompass correlated variables, such as surgeon volume, transplant center status, teaching hospital status, number of beds, etc. This inconsistency, coupled with varying recommendations, does not provide a sufficient evidence base to justify a change in the current volume criteria specified in the current NCD.
VIII. CMS Analysis
Introduction: National coverage determinations are determinations by the Secretary with respect to whether or not a particular item or service is covered nationally under title XVIII of the Social Security Act. § 1862(l)(6), 1869(f)(1)(B). Among other things, in order to be covered by Medicare, an item or service must fall within one or more benefit categories contained within Part A or Part B, and must not be otherwise excluded from coverage. Moreover, with limited exceptions, items or services must be reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. §1862(a)(1)(A).
When making national coverage determinations, we evaluate the evidence related to our analytic questions based on the quality, strength and totality of evidence presented in the reviewed literature. As part of this evaluation, it is important to consider whether the evidence is relevant to the Medicare beneficiary population. In determining the generalizability of the results of the body of evidence to the Medicare population, we consider, at minimum, the age, race and gender of the study participants.
Artificial Hearts:
For this artificial heart NCD reconsideration, CMS focused on the following question:
Q1: Is the evidence sufficient to end coverage with evidence development for artificial hearts?
Based on the totality of the limited evidence generated through coverage with evidence development and peer-reviewed articles and varying patient characteristics, CMS finds that a national coverage determination for artificial hearts is no longer necessary or appropriate and will allow MACs to determine coverage of artificial hearts in particular cases. Research on artificial hearts has shown improved survival for patients with severe biventricular failure. For certain patients, an artificial heart may be reasonable and necessary to treat the patient’s heart disease. However, due to the very low number of procedures, < 1 % of the Medicare population, and the need for careful patient selection, CMS believes coverage of artificial hearts is an appropriate determination made by the Medicare Administrative Contractors (MACs).
The MACs are structured to be able to take into account a beneficiary’s particular clinical circumstances which are especially important when overall prevalence is very low. Thus, we are finalizing as proposed and removing our national coverage determination at § 20.9. In the absence of a national coverage determination, MACs would make the coverage determination under § 1862(a)(1)(A) of the Social Security Act.
Left Ventricular Assist Devices (LVADs):
For this LVAD NCD reconsideration, CMS focused on the following questions:
Q1: Is the evidence sufficient to conclude that modifying the current patient selection criteria for LVAD implantation will improve health outcomes for Medicare beneficiaries?
Q2: Is the evidence sufficient to conclude that modifying the current facility criteria for LVAD implantation will improve health outcomes for Medicare beneficiaries?
Is the evidence sufficient to conclude that modifying the current patient selection criteria for LVAD implantation will improve health outcomes for Medicare beneficiaries?
With respect to LVADS, this decision is limited to durable, intracorporeal, LVADs, and does not include temporary VADs or extracorporeal membrane oxygen (ECMO). The evidence is adequate to assess which patient selection criteria predict the most successful patient outcomes. We are updating the final coverage criteria to align with patient inclusion criteria derived from large, randomized controlled trials. This will expand coverage to a greater number of LVAD candidates who are likely to benefit from LVAD use while maintaining an adequate safety profile. This is consistent with the majority of public commenters who supported expanded coverage.
Quality and Strength of Evidence
This reconsideration assesses patient selection criteria as informed by two major randomized trials and eight observational studies that have been conducted since the last reconsideration in 2013:
- ENDURANCE trial (Rogers et al., 2017), sample size=446.
- Multicenter Study of MagLev Technology in Patients Undergoing Mechanical Circulatory Support Therapy with HeartMate 3 trial (MOMENTUM 3) (Mehra et al., 2019), sample size=1028.
A side-by-side comparison of the trial patient inclusion criteria appears in the table below. The patient populations in the two trials (ENDURANCE & MOMENTUM 3) were comprised of LVAD candidates who derived documented efficacy from LVAD utilization while maintaining an adequate safety profile; therefore, the inclusion and exclusion criteria from these trials serve as a foundation for updating patient selection criteria.
Table 5. Inclusion criteria from major randomized LVAD trials (since 2013)
| |
ENDURANCE |
MOMENTUM 3 |
| Author, Year |
Rogers, 2017 |
Mehra, 2019 |
| Patients Included |
Ineligible for transplant (DT) NYHA Class IIIB or IV LVEF ≤ 25% On OMM, including dietary salt restriction and diuretics, 45 of the last 60 days and failing to respond; or Advanced HF for at least 14 days, and dependent on IABP for 7 days and/or inotropes for at least 14 days |
DT, BTT, BTC NYHA Class III with dyspnea upon mild physical activity, or NYHA Class IV LVEF ≤ 25% Inotrope dependent; or Not inotrope dependent, but CI < 2.2 L/min/m2, and either: - on OMM 45 of last 60 days and failing to respond; or
- advanced HF for at least 14 days and dependent on IABP for at least 7 days |
MOMENTUM 3, the largest and most recent LVAD trial, randomized 1028 patients with advanced heart failure to either a centrifugal-flow pump (HeartMate 3) or to an axial-flow pump (HeartMate II), irrespective of therapeutic intent (bridge to transplantation, bridge to candidacy, or destination therapy) at the time of implant. Both groups received optimal medical management, including antithrombotic therapy. The trial was designed specifically to conduct subgroup analyses comparing device results across the BTT/BTC and DT cohorts.
Of the two trials, we are aligning patient selection criteria for this NCD reconsideration with MOMENTUM 3’s major inclusion criteria. MOMENTUM 3 was a larger randomized trial, with broader inclusion criteria regardless of therapeutic intent categorizations (BTT, BTC, DT). The study was methodologically strong and generated improved results of survival at two years free of disabling stroke and reoperation. The patient selection criteria are as follows:
Patient Selection Criteria
Based on the available evidence, we will cover left ventricular assist devices (LVADs) if they are FDA approved for short-term (e.g., bridge-to-transplant) or long-term (e.g., destination therapy) mechanical circulatory support for heart failure patients who meet the following criteria:
- Have New York Heart Association (NYHA) Class IV heart failure; and
- Have a left ventricular ejection fraction (LVEF) ≤ 25%; and
- Are inotrope dependent>
OR
have a Cardiac Index (CI) < 2.2 L/min/m2, while not on inotropes, and also meet one of the following:
- Are on optimal medical management (OMM), based on current heart failure practice guidelines for at least 45 out of the last 60 days and are failing to respond; or
- Have advanced heart failure for at least 14 days and are dependent on an intra‐aortic balloon pump (IABP) or similar temporary mechanical circulatory support for at least 7 days.
While the MOMENTUM 3 study inclusion criteria encompassed patients with NYHA class Ⅲ heart failure, none of their interim or final reports (Mehra et al., 2018; Mehra et al., 2017; Mehra et al., 2019) the trial supplements, or the secondary analysis of trial data (Goldstein et al., 2020) reported the number of NYHA class Ⅲ patients in the trial. Therefore, the available evidence is not sufficient to change the previous requirement of NYHA class Ⅳ classification.
The pre-specified subgroup analysis of therapeutic intent in the MOMENTUM 3 trial found that the therapeutic intent category (i.e., BTT, DT or BTC) did not affect the association between the device and the primary outcome of survival at 2 years free of disabling stroke and reoperation (Goldstein et al., 2020):
A superior effect on outcomes of HeartMate 3 over HeartMate II was similar in BTT and DT subgroups, and
In HeartMate 3 patients, there was no difference in event-free survival between BTT and DT
groups.
The Goldstein subgroup analysis of the MOMENTUM 3 trial data foundthat positive health outcomes do not depend on whether a patient is characterized as DT or BTT or BTC at the time of implantation. Patients move dynamically between these categories.
In the MOMENTUM 3 trial, approximately 12% of patients receiving a HeartMate 3 pump who were initially categorized as destination therapy ended up receiving a heart transplant within 2 years. Forty-three percent of patients with this device who were initially categorized as bridge to transplant or bridge to candidacy remained on the device at the 2-year mark. A European registry study demonstrated a similar pattern, stating that although BTT is the device strategy in the majority of LT-MCS (long-term mechanical circulatory support) recipients, only a minority receive a donor organ in Europe. In another recent European report of the ELEVATE (Evaluating the HeartMate 3 with Full MagLev Technology in a Post-Market Approval Setting) registry of more than 450 patients with LT-MCS, only 2% received a transplant after 1 year, despite 26% of the patients receiving an implant as a DT strategy (Morshuis et al., 2018; Potapov et al., 2019).
The European Association for Cardio-Thoracic Surgery expert consensus on long-term mechanical circulatory support also stated that they generally refrained from using the designations of bridge to transplant and destination therapy in accordance with the MOMENTUM 3 trial (Potapov et al., 2019).
Like the 2018 Intermacs annual report (Kormos 2019), the 2019 report’s subgroup analysis found that survival differed depending on therapeutic intent: “Those whose LVAD was implanted as BTT have superior survival compared with BTC and DT patients” (Teuteberg 2020). This conflicts with the pre-specified subgroup analysis of the MOMENTUM 3 study, which found no significant difference between therapeutic intent subgroups (Goldstein 2020).
This difference between the trial and registry study might be explained in part by size and/or setting. The registry study included over 13,000 patients representing LVAD implantation in broad community practice. MOMENTUM 3 included a little over 1,000 patients representing LVAD implantation in a rigorous clinical trial. Larger sample sizes might demonstrate that DT patients fare worse than BTT patients regardless of treatment setting because they are generally sicker; that is why they were not considered surgical candidates in the first place. A significant portion of these DT patients may eventually become surgical candidates, as demonstrated by MOMENTUM 3 and other studies.
The MOMENTUM 3 trial data confirmed and echoed previous literature questioning the clinical relevance of characterizing patients pre-implant with therapeutic intent categories of BTT, BTC, or DT. Patients often transition between device strategies following device implant. Therefore, based on the totality of the evidence including the MOMENTUM 3 trial, the subgroup analysis, and the consensus statement, we are modifying the NCD to update the patient selection criteria irrespective of therapeutic intent category. We believe these changes will facilitate better access and clinical care regardless of whether patients start in one therapeutic intent category or another, or dynamically move between them.
Is the evidence sufficient to conclude that modifying the current facility criteria for LVAD implantation will improve health outcomes for Medicare beneficiaries?
Removing the therapeutic intent categories inherently leads to the need to reconsider the current facility criteria established distinctly for DT and BTT.
There has not been sufficient evidence published since the last reconsideration of this policy in 2013 to support a change to most of the parameters in the existing facility requirements. A number of studies have provided data on volume, yet this newer evidence as a whole has been inconclusive. While these studies are generally supportive of maintaining a volume requirement, the threshold for volume and evidence characterizing volume varies from study to study. Given this general support, yet corresponding differences in the evidence, we are finalizing as proposed and will maintaining the existing facility volume requirements while removing the therapeutic intent categories of DT and BTT.
Quality and Strength of Evidence
The 2013 NCD required specific expertise for the team implanting VADs for DT. One requirement was that the team include at least “one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 durable, intracorporeal, left VADs as BTT or DT over the course of the previous 36 months with activity in the last year.”
Several studies evaluated LVAD volume as a predictor of mortality, but did not standardize these volume assessments. Some evaluated surgeon volume as distinct from hospital volume. Others used hospital volume as a broad measure that might encompass correlated variables, such as surgeon volume, transplant center status, teaching hospital status, number of beds, etc. This inconsistency, coupled with varying recommendations, does not provide a sufficient evidence base to justify a change in the current volume criteria specified in the current NCD. While we are not making any changes to these requirements, the pertinent evidence is summarized below.
A large, observational study of 7714 patients undergoing LVAD implantation between 2007 and 2012 at 88 hospitals (581 surgeons), found that surgeons’ LVAD procedure volume, not annual hospital procedure volume, was associated with in-hospital mortality (Davis et al., 2016). The study results suggest that the critical threshold for surgeons’ LVAD procedure volume is 12 LVAD implantations per year.
A second observational study of 1749 LVAD implants between 2008 and 2011, found that, “High annual LVAD volume is associated with significantly lower in-hospital mortality and length of stay post-LVAD implantation, with an optimal volume threshold of >20 LVADs/y, suggesting that center volume is crucial for better care of these patients” (Shah et al., 2016). The investigators did not assess surgeon volume as distinct from hospital volume. The authors stated that: “The most important finding of our study is identification of an optimal volume threshold of 20 LVADs/ year, associated with <10% in-hospital mortality. The lack of benefit beyond 20 LVADs/year may be because very high-volume centers are major tertiary care referral centers, caring for critically ill advanced heart failure patients, and any further mortality benefit is offset by the critical illness of these patients.” It is unknown to what extent the association of hospital volume might be driven by surgeon volume.
A larger and more recent study based on 7416 patients in the INTERMACS registry undergoing LVAD implantation between 2009 and 2015 assessed the impact of center volume on survival (Cowger et al., 2017). The study found that patients undergoing LVAD implant at very low (<10 implants per year) and high volume U.S. centers (>50 implants per year) have poorer outcomes when compared to patients having surgery at centers performing 31 to 50 VADs a year. The study did not assess surgeon volume independently. The investigators acknowledge that: “Unmeasured factors, such as surgical experience, perioperative management, quality and frequency of outpatient follow-up, identification and
management of LVAD-related complications, and variations in patient management protocols (e.g., anticoagulation and blood pressure) could also be hypothesized to play a role in the increased incidence of death at very low volume LVAD centers. Certainly, more studies are needed to better understand the causes of increased mortality at low volume centers.” The authors also commented on the increased mortality found with very high hospital volumes: “While the higher mortality observed at higher volume centers may be due to referral bias, these findings support the need for regular performance improvement evaluations, comparing individual center outcomes to risk-adjusted national averages. Coincidently, strategies enacted to improve average LVAD patient survival in those centers that underperform are warranted. Finally, INTERMACS data would suggest that current U.S. VAD center standards (10 VAD/total artificial hearts over 3 years) imposed by CMS are too lenient and warrant reconsideration.”
A study conducted in adult recipients of a primary, continuous-flow LVAD as destination therapy between January 2012 and March 2014 from INTERMACS classified patients by implanting center as transplant (n=3323) or nontransplant (n=260) (Brinkley et al., 2018). The investigators used a cut point of < 15 or ≥ 15 procedures to represent baseline volume in 2011. The investigators did not assess surgeon volume distinctly but reported that, “Cumulative LVAD volume ≥15 was also not associated with improved mortality (HR, 0.88 [95% CI, 0.68–1.15]; P=0.35). Because the majority of transplant centers had higher volumes than nontransplant centers, a subgroup analysis compared patients implanted at centers with similar volume. Within the cohort limited to centers with volume <21 primary implants in 2011, 198 patients were from nontransplant centers and 1206 from transplant centers. Neither patient survival (log-rank P=0.22) nor freedom from death or major adverse event (log-rank P=0.84) was different between these groups. Twelve-month survival was 71.8% (64.9%–78.6%) at nontransplant centers compared with 76.6% (74%–79.2%) at transplant centers, whereas freedom from death or major adverse event was 29.8% (22.9%–36.7%) versus 29.9% (27.1%–32.7%), respectively.”
Therefore, while the data are mixed, most studies found a decrease in mortality with a moderate increase in volume; however, caution is needed in interpretation as hospital volume may be a proxy for other factors, such as surgeon volume. Additional research to sort out the complexity of the volume measurement and ideal volume threshold would be needed for us to make changes to the current physician volume requirements. To make such a change, there must be strong evidence that higher center or physician volume requirements would improve patient health outcomes for Medicare beneficiaries receiving a VAD
Facility Criteria
For DT, beneficiaries receiving a VAD must currently be managed by an explicitly identified, cohesive, multidisciplinary team of medical professionals with the appropriate qualifications, training, and experience. Additionally, DT facilities must currently be credentialed by an organization approved by CMS. The 2013 NCD maintained this requirement because advanced heart failure patients and LVAD recipients are medically fragile, and the procedure is high risk and technically complex. There also were no established consensus-based standards for facility requirements at the time of the last reconsideration. Since our last reconsideration in 2013, the new evidence is supportive of a volume requirement but is too inconsistent to warrant a change to the current facility criteria. To ensure consistent quality of care for Medicare beneficiaries, facilities implanting LVADs for short-term or long-term mechanical circulatory support in patients with advanced heart failure must continue to meet the professional team and credentialing criteria. These criteria remain unchanged from the current NCD and we do not believe this will limit appropriate access to LVADs for beneficiaries.
Health Disparities
Gender and race: Heart failure disproportionately affects racial/ethnic minorities and women. African Americans are more likely to have heart failure than people of other races, with a high prevalence at an early age (Sharma, Colvin-Adams, & Yancy, 2014). Incidence rates of heart failure in men approximately double with each 10-year increase in age from 65 to 85 years; however, the heart failure incidence rate triples for women between ages 65 to 74 and 75 to 84 years (Mozaffarian et al., 2016). According to the 2019 INTERMACS Database Annual Report, women continued to only represent approximately 20% of continuous flow LVAD implants, and approximately 32.8% of LVAD recipients were non-white (Kormos et al., 2019).
Approximately 81% of the MOMENTUM 3 trial participants were male; 69% were white, 26% black, 1.1% Asian, 0.8% Native Hawaiian or Pacific Islander and 3.8% other. The study reported that no interaction between the groups was observed for the prespecified subgroups of age, sex, race or ethnic group, intended goal of pump support (bridge to transplantation or destination therapy), or INTERMACS profile with regard to the primary end point. The ENDURANCE study patients were approximately 79.4% male; 77% white, 22% black, and 1% other. The trial was unlikely powered to perform a subgroup analysis by sex and race.
The evidence base for treatment of heart failure in women is not robustly supported by sex-specific data (Taylor, 2015). CMS is interested in broader evidence on characteristics predicting successful outcomes, including the influence of sex and race. In addition, CMS is interested in information on access to LVAD by race to assess whether unintended barriers might preclude access for minority populations among Medicare beneficiaries.
IX. Conclusion
A. The Centers for Medicare & Medicaid Services (CMS) is making changes to two separate, but medically related, National Coverage Determinations (NCDs). Given new evidence in the peer-reviewed medical literature, we are removing the NCD for Artificial Hearts and Related Devices established at §20.9 of the Medicare National Coverage Determination Manual. We are also revising the NCD at § 20.9.1 that provides coverage for Ventricular Assist Devices (VADs) for Bridge-to-Transplant and Destination Therapy. We summarize the changes below and fully explain the changes in the Analysis section of this NCD decision memo.
Artificial Hearts
CMS is removing the NCD at § 20.9, ending coverage with evidence development for artificial hearts and permitting Medicare coverage determinations for artificial hearts to be made by the Medicare Administrative Contractors (MACs) under § 1862(a)(1)(A) of the Social Security Act.
Ventricular Assist Devices
The decision is limited to durable, intracorporeal, left ventricular assist devices (LVADs), and does not include temporary VADs or extracorporeal membrane oxygen (ECMO). CMS is establishing coverage according to the following conditions:
Patient Selection Criteria
- Removes the current therapeutic intent-to-treat criteria of bridge-to-transplant (BTT) and destination therapy (DT), by removing the following BTT requirements:
- The requirement that the patient is active on the waitlist maintained by the Organ Procurement and Transplantation Network (OPTN).
- The requirement that the implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved transplant center prior to implantation of the VAD.
- Extends evidence based patient selection criteria, that previously applied only to DT, for all LVAD procedures for short-term (e.g., bridge-to-recovery and bridge-to-transplant) or long-term (e.g., destination therapy) mechanical circulatory support (see below in covered indications).
- Modifies patient selection criteria to reflect the major inclusion criteria of contemporary trials.
Facility criteria
- The facility criteria remain unchanged from the current NCD. The removal of therapeutic intent is not expected to decrease appropriate patient access to LVADs.
Below is the 20.9.1 NCD (Ventricular Assist Devices). All proposed changes are in italics. Any part of 20.9.1 that is not italicized is not proposed to be revised.
B. Covered Indications
1. Advanced Heart Failure
Left ventricular assist devices (LVADs) are covered if they are FDA approved for short-term (e.g.,
bridge-to-recovery and bridge-to-transplant) or long-term (e.g., destination therapy) mechanical circulatory support for heart failure patients who meet the following criteria:
- Have New York Heart Association (NYHA) Class IV heart failure; and
- Have a left ventricular ejection fraction (LVEF) ≤ 25%; and
- Are inotrope dependent
OR
have a Cardiac Index (CI) < 2.2 L/min/m2, while not on inotropes, and also meet one of the following:
- Are on optimal medical management (OMM), based on current heart failure practice guidelines for at least 45 out of the last 60 days and are failing to respond; or
- Have advanced heart failure for at least 14 days and are dependent on an intra‐aortic balloon pump (IABP) or similar temporary mechanical circulatory support for at least 7 days.
Beneficiaries receiving a VAD must be managed by an explicitly identified, cohesive, multidisciplinary team of medical professionals with appropriate qualifications, training, and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in informed decision making. The team members must be based at the facility and must include individuals with experience working with patients before and after placement of a VAD.
The team must include, at a minimum:
- At least one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 durable, intracorporeal, left ventricular assist devices over the course of the previous 36 months with activity in the last year.
- At least one cardiologist trained in advanced heart failure with clinical competence in medical- and device-based management including VADs, and clinical competence in the management of patients before and after placement of a VAD.
- A VAD program coordinator.
- A social worker.
- A palliative care specialist.
The process for organizations to apply for CMS approval to be designated as a credentialing organization for VAD facilities is posted on our web site along with a list of approved credentialing organizations, approved standard versions, and credentialed facilities: http://www.cms.gov/Medicare/Medicare-General-Information/MedicareApprovedFacilitie/VAD-Destination-Therapy-Facilities.html
See Appendix B for the manual language.
APPENDIX A
General Methodological Principles of Study Design
(Section VI of the Decision Memorandum)
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service is reasonable and necessary. The overall objective for the critical appraisal of the evidence is to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for patients.
We divide the assessment of clinical evidence into three stages: 1) the quality of the individual studies; 2) the generalizability of findings from individual studies to the Medicare population; and 3) overarching conclusions that can be drawn from the body of the evidence on the direction and magnitude of the intervention’s potential risks and benefits.
The methodological principles described below represent a broad discussion of the issues we consider when reviewing clinical evidence. However, it should be noted that each coverage determination has its unique methodological aspects.
Assessing Individual Studies
Methodologists have developed criteria to determine weaknesses and strengths of clinical research. Strength of evidence generally refers to: 1) the scientific validity underlying study findings regarding causal relationships between health care interventions and health outcomes; and 2) the reduction of bias. In general, some of the methodological attributes associated with stronger evidence include those listed below:
- Use of randomization (allocation of patients to either intervention or control group) in order to minimize bias.
- Use of contemporaneous control groups (rather than historical controls) in order to ensure comparability between the intervention and control groups.
- Prospective (rather than retrospective) studies to ensure a more thorough and systematical assessment of factors related to outcomes.
- Larger sample sizes in studies to demonstrate both statistically significant as well as clinically significant outcomes that can be extrapolated to the Medicare
population. Sample size should be large enough to make chance an unlikely explanation for what was found.
- Masking (blinding) to ensure patients and investigators do not know to that group patients were assigned (intervention or control). This is important especially in subjective outcomes, such as pain or quality of life, where enthusiasm and psychological factors may lead to an improved perceived outcome by either the patient or assessor.
Regardless of whether the design of a study is a randomized controlled trial, a non-randomized controlled trial, a cohort study or a case-control study, the primary criterion for methodological strength or quality is to the extent that differences between intervention and control groups can be attributed to the intervention studied. This is known as internal validity. Various types of bias can undermine internal validity. These include:
- Different characteristics between patients participating and those theoretically eligible for study but not participating (selection bias).
- Co-interventions or provision of care apart from the intervention under evaluation (performance bias).
- Differential assessment of outcome (detection bias).
- Occurrence and reporting of patients who do not complete the study (attrition bias).
In principle, rankings of research design have been based on the ability of each study design category to minimize these biases. A randomized controlled trial minimizes systematic bias (in theory) by selecting a sample of participants from a particular population and allocating them randomly to the intervention and control groups. Thus, in general, randomized controlled studies have been typically assigned the greatest strength, followed by non-randomized clinical trials and controlled observational studies. The design, conduct and analysis of trials are important factors as well. For example, a well-designed and conducted observational study with a large sample size may provide stronger evidence than a poorly designed and conducted randomized controlled trial with a small sample size. The following is a representative list of study designs (some of that have alternative names) ranked from most to least methodologically rigorous in their potential ability to minimize systematic bias:
Randomized controlled trials
Non-randomized controlled trials
Prospective cohort studies
Retrospective case control studies
Cross-sectional studies
Surveillance studies (e. g. , using registries or surveys)
Consecutive case series
Single case reports
When there are merely associations but not causal relationships between a study’s variables and outcomes, it is important not to draw causal inferences. Confounding refers to independent variables that systematically vary with the causal variable. This distorts measurement of the outcome of interest because its effect size is mixed with the effects of other extraneous factors. For observational, and in some cases randomized controlled trials, the method in that confounding factors are handled (either through stratification or appropriate statistical modeling) are of particular concern. For example, in order to interpret and generalize conclusions to our population of Medicare patients, it may be necessary for studies to match or stratify their intervention and control groups by patient age or co-morbidities.
Methodological strength is, therefore, a multidimensional concept that relates to the design, implementation and analysis of a clinical study. In addition, thorough documentation of the conduct of the research, particularly study selection criteria, rate of attrition and process for data collection, is essential for CMS to adequately assess and consider the evidence.
Generalizability of Clinical Evidence to the Medicare Population
The applicability of the results of a study to other populations, settings, treatment regimens and outcomes assessed is known as external validity. Even well-designed and well-conducted trials may not supply the evidence needed if the results of a study are not applicable to the Medicare population. Evidence that provides accurate information about a population or setting not well represented in the Medicare program would be considered but would suffer from limited generalizability.
The extent to that the results of a trial are applicable to other circumstances is often a matter of judgment that depends on specific study characteristics, primarily the patient population studied (age, sex, severity of disease and presence of co-morbidities) and the care setting (primary to tertiary level of care, as well as the experience and specialization of the care provider). Additional relevant variables are treatment regimens (dosage, timing and route of administration), co-interventions or concomitant therapies, and type of outcome and length of follow-up.
The level of care and the experience of the providers in the study are other crucial elements in assessing a study’s external validity. Trial participants in an academic medical center may receive more or different attention than is typically available in non-tertiary settings. For example, an investigator’s lengthy and detailed explanations of the potential benefits of the intervention and/or the use of new equipment provided to the academic center by the study sponsor may raise doubts about the applicability of study findings to community practice.
Given the evidence available in the research literature, some degree of generalization about an intervention’s potential benefits and harms is invariably required in making coverage determinations for the Medicare population. Conditions that assist us in making reasonable generalizations are biologic plausibility, similarities between the populations studied and Medicare patients (age, sex, ethnicity and clinical presentation) and similarities of the intervention studied to those that would be routinely available in community practice.
A study’s selected outcomes are an important consideration in generalizing available clinical evidence to Medicare coverage determinations. One of the goals of our determination process is to assess health outcomes. These outcomes include resultant risks and benefits such as increased or decreased morbidity and mortality. In order to make this determination, it is often necessary to evaluate whether the strength of the evidence is adequate to draw conclusions about the direction and magnitude of each individual outcome relevant to the intervention under study. In addition, it is important that an intervention’s benefits are clinically significant and durable, rather than marginal or short-lived. Generally, an intervention is not reasonable and necessary if its risks
outweigh its benefits.
If key health outcomes have not been studied or the direction of clinical effect is inconclusive, we may also evaluate the strength and adequacy of indirect evidence linking intermediate or surrogate outcomes to our outcomes of interest.
Assessing the Relative Magnitude of Risks and Benefits
Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits. Health outcomes are one of several considerations in determining whether an item or service is reasonable and necessary. CMS places greater emphasis on health outcomes actually experienced by patients, such as quality of life, functional status, duration of disability, morbidity and mortality, and less emphasis on outcomes that patients do not directly experience, such as intermediate outcomes, surrogate outcomes, and laboratory or radiographic responses. The direction, magnitude, and consistency of the risks and benefits across studies are also important considerations. Based on the analysis of the strength of the evidence, CMS assesses the relative magnitude of an intervention or technology’s benefits and risk of harm to Medicare beneficiaries.
APPENDIX B
Medicare National Coverage Determinations Manual
This draft NCD is subject to formal revisions and formatting changes prior to the release of the final NCD contractor instructions and publication in the Medicare National Coverage Determinations Manual.
Table of Contents
(Rev.)
20.9 – Artificial Hearts and Related Devices
Effective November XX, 2020, Artificial Hearts has been removed from the NCD Manual.
20.9.1 – Ventricular Assist Devices (VADs)
A. General
A ventricular assist device (VAD) is surgically attached to one or both intact ventricles and is used to assist or augment the ability of a damaged or weakened native heart to pump blood. Improvement in the performance of the native heart may allow the device to be removed.
Indications and Limitations of Coverage
B. Nationally Covered Indications
1. Post-cardiotomy (effective for services performed on or after October 18, 1993)
Post-cardiotomy is the period following open-heart surgery. VADs used for support of blood circulation post-cardiotomy are covered only if they have received approval from the Food and Drug Administration (FDA) for that purpose, and the VADs are used according to the FDA-approved labeling instructions.
2. Left ventricular assist devices (LVADs) are covered if they are FDA approved for short-term (e.g., bridge-to-recovery and bridge-to-transplant) or long-term (e.g., destination therapy) mechanical circulatory support for heart failure patients who meet the following criteria:
- Have New York Heart Association (NYHA) Class IV heart failure; and
- Have a left ventricular ejection fraction (LVEF) ≤ 25%; and
- Are inotrope dependent
OR
have a Cardiac Index (CI) < 2.2 L/min/m2, while not on inotropes, and also meet one of the following:
- Are on optimal medical management (OMM), based on current heart failure practice guidelines for at least 45 out of the last 60 days and are failing to respond; or
- Have advanced heart failure for at least 14 days and are dependent on an intra‐aortic balloon pump (IABP) or similar temporary mechanical circulatory support for at least 7 days.
Beneficiaries receiving a VAD must be managed by an explicitly identified, cohesive, multidisciplinary team of medical professionals with appropriate qualifications, training, and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in informed decision making. The team members must be based at the facility and must include individuals with experience working with patients before and after placement of a VAD.
The team must include, at a minimum:
- At least one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 durable, intracorporeal, left ventricular assist devices over the course of the previous 36 months with activity in the last year.
- At least one cardiologist trained in advanced heart failure with clinical competence in medical- and device-based management including VADs, and clinical competence in the
management of patients before and after placement of a VAD.
- A VAD program coordinator.
- A social worker.
- A palliative care specialist.
Facilities must be credentialed by an organization approved by CMS. The process for organizations to apply for CMS approval to be designated as a credentialing organization for LVAD facilities is posted on our web site along with a list of approved credentialing organizations, approved standard versions, and credentialed facilities: http://www.cms.gov/Medicare/Medicare-General-Information/MedicareApprovedFacilitie/VAD-Destination-Therapy-Facilities.html
C. Nationally Non-Covered Indications
All other indications for the use of VADs not otherwise listed remain non-covered, except in the context of Category B investigational device exemption clinical trials (42 CFR 405) or as a routine cost in clinical trials defined under section 310.1 of the National Coverage Determinations (NCD) Manual.
D. Other
This policy does not address coverage of VADs for right ventricular support, biventricular support, use in beneficiaries under the age of 18, use in beneficiaries with complex congenital heart disease, or use in beneficiaries with acute heart failure without a history of chronic heart failure. Coverage under section 1862(a)(1)(A) of the Act for VADs in these situations will be made by local Medicare Administrative Contractors within their respective jurisdictions.
APPENDIX C – NCD 20.9 (2013)
A. General
An artificial heart is a biventricular replacement device which requires removal of a substantial part of the native heart, including both ventricles. Removal of this device is not compatible with life, unless the patient has a heart transplant.
Indications and Limitations of Coverage
B. Nationally Covered Indications
1. Bridge-to-transplant (BTT) (effective for services performed on or after May 1, 2008)
An artificial heart for bridge-to-transplantation (BTT) is covered when performed under coverage with evidence development (CED) when a clinical study meets all of the criteria listed below. The clinical study must address at least one of the following questions:
- Were there unique circumstances such as expertise available in a particular facility or an unusual combination of conditions in particular patients that affected their outcomes?
- What will be the average time to device failure when the device is made available to larger numbers of patients?
- Do results adequately give a reasonable indication of the full range of outcomes (both positive and negative) that might be expected from more widespread use?
The clinical study must meet all of the criteria stated in Section D of this policy. The above information should be mailed to: Director, Coverage and Analysis Group, Centers for Medicare & Medicaid Services (CMS), Re: Artificial Heart, Mailstop S3-02-01, 7500 Security Blvd, Baltimore, MD 21244-1850.
Clinical studies that are determined by CMS to meet the above requirements will be listed on the CMS Web site at: http://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development/Artificial-Hearts.html.
2. Destination therapy (DT) (effective for services performed on or after May 1, 2008)
An artificial heart for destination therapy (DT) is covered when performed under CED when a clinical study meets all of the criteria listed below. The clinical study must address at least one of the following questions:
- Were there unique circumstances such as expertise available in a particular facility or an unusual combination of conditions in particular patients that affected their outcomes?
- What will be the average time to device failure when the device is made available to larger numbers of patients?
- Do results adequately give a reasonable indication of the full range of outcomes (both positive and negative) that might be expected from more widespread use?
The clinical study must meet all of the criteria stated in Section D of this policy. The above information should be mailed to: Director, Coverage and Analysis Group, Centers for Medicare & Medicaid Services, Re: Artificial Heart, Mailstop S3-02-01, 7500 Security Blvd, Baltimore, MD 21244-1850.
Clinical studies that are determined by CMS to meet the above requirements will be listed on the CMS Web site at: http://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development/Artificial-Hearts.html.
C. Nationally Non-Covered Indications
All other indications for the use of artificial hearts not otherwise listed remain non-covered, except in the context of Category B investigational device exemption clinical trials (42 CFR 405) or as a routine cost in clinical trials defined under section 310.1 of the National Coverage Determinations (NCD) Manual.
D. Other
Clinical study criteria:
- The study must be reviewed and approved by the Food and Drug Administration (FDA).
- The principal purpose of the research study is to test whether a particular intervention potentially improves the participants' health outcomes.
- The research study is well supported by available scientific and medical information, or it is intended to clarify or establish the health outcomes of interventions already in common clinical use.
- The research study does not unjustifiably duplicate existing studies.
- The research study design is appropriate to answer the research question being asked in the study.
- The research study is sponsored by an organization or individual capable of executing the proposed study successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found at 45 CFR Part 46. If a study is FDA-regulated it also must be in compliance with 21 CFR Parts 50 and 56.
- All aspects of the research study are conducted according to appropriate standards of scientific integrity (see http://www.icmje.org).
- The research study has a written protocol that clearly addresses, or incorporates by reference, the standards listed here as Medicare requirements for CED.
- The clinical research study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Trials of all medical technologies measuring therapeutic outcomes as one of the objectives meet this standard only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research study is registered on the www.ClinicalTrials.gov Web site by the principal sponsor/investigator as demonstrated by having a Clinicaltrials.gov Identifier.
- The research study protocol specifies the method and timing of public release of all pre-specified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 24 months of the end of data collection. If a report is planned to be published in a peer-reviewed journal, then that initial release may be an abstract that meets the requirements of the International Committee of Medical Journal Editors (ICMJE) (http://www.icmje.org). However a full report of the outcomes must be made public no later than three (3) years after the end of data collection.
- The research study protocol must explicitly discuss subpopulations affected by the treatment under investigation, particularly traditionally under-represented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations in the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of under-represented populations, the protocol must discuss why these criteria are necessary.
- The research study protocol explicitly discusses how the results are or are not expected to be generalizable to the Medicare population to infer whether Medicare patients may benefit from the intervention. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability, or Medicaid eligibility.
Consistent with section 1142 of the Social Security Act (the Act), the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions.
The principal investigator of an artificial heart clinical study seeking Medicare payment should submit the following documentation to CMS and should expect to be notified when the CMS review is complete:
- Complete study protocol (must be dated or identified with a version number);
- Protocol summary;
- Statement that the submitted protocol version has been agreed upon by the FDA;
- Statement that the above study standards are met;
- Statement that the study addresses at least one of the above questions related to artificial hearts;
- Complete contact information (phone number, email address, and mailing address); and,
- Clinicaltrials.gov Identifier.
APPENDIX D – NCD 20.9.1 (2013)
A. General
A ventricular assist device (VAD) is surgically attached to one or both intact ventricles and is used to assist or augment the ability of a damaged or weakened native heart to pump blood. Improvement in the performance of the native heart may allow the device to be removed.
Indications and Limitations of Coverage
B. Nationally Covered Indications
1. Post-cardiotomy (effective for services performed on or after October 18, 1993)
Post-cardiotomy is the period following open-heart surgery. VADs used for support of blood circulation post-cardiotomy are covered only if they have received approval from the Food and Drug Administration (FDA) for that purpose, and the VADs are used according to the FDA-approved labeling instructions.
2. Bridge-to-Transplant (effective for services performed on or after January 22, 1996)
The VADs used for bridge to transplant are covered only if they have received approval from the FDA for that purpose, and the VADs are used according to FDA-approved labeling instructions. All of the following criteria must be fulfilled in order for Medicare coverage to be provided for a VAD used as a bridge to transplant:
- The patient is approved for heart transplantation by a Medicare-approved heart transplant center and is active on the Organ Procurement and Transplantation Network (OPTN) heart transplant waitlist.
- The implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved transplant center under which the patient is listed prior to implantation of the VAD.
3. Destination Therapy (DT) (effective for services performed on or after October 1, 2003)
Destination therapy (DT) is for patients that require mechanical cardiac support. The VADs used for DT are covered only if they have received approval from the FDA for that purpose.
Patient Selection (effective November 9, 2010):
The VADs are covered for patients who have chronic end-stage heart failure (New York Heart Association Class IV end-stage left ventricular failure) who are not candidates for heart transplantation at the time of VAD implant, and meet the following conditions:
- Have failed to respond to optimal medical management (including beta-blockers and ACE inhibitors if tolerated) for 45 of the last 60 days, or have been balloon pump-dependent for 7 days, or IV inotrope-dependent for 14 days; and,
- Have a left ventricular ejection fraction (LVEF) < 25%; and,
- Have demonstrated functional limitation with a peak oxygen consumption of ≤ 14 ml/kg/min unless balloon pump- or inotrope-dependent or physically unable to perform the test.
Facility Criteria (effective October 30, 2013):
Facilities currently credentialed by the Joint Commission for placement of VADs as DT may continue as Medicare-approved facilities until October 30, 2014. At the conclusion of this transition period, these facilities must be in compliance with the following criteria as determined by a credentialing organization. As of the effective date, new facilities must meet the following criteria as a condition of coverage of this procedure as DT under section 1862(a)(1)(A) of the Social Security Act (the Act):
Beneficiaries receiving VADs for DT must be managed by an explicitly identified cohesive, multidisciplinary team of medical professionals with the appropriate qualifications, training, and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in informed decision making. The team members must be based at the facility and must include individuals with experience working with patients before and after placement of a VAD.
The team must include, at a minimum:
- At least one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 durable, intracorporeal, left VADs as BTT or DT over the course of the previous 36 months with activity in the last year.
- At least one cardiologist trained in advanced heart failure with clinical competence in medical and device-based management including VADs, and clinical competence in the management of patients before and after heart transplant.
- A VAD program coordinator.
- A social worker.
- A palliative care specialist.
Facilities must be credentialed by an organization approved by the Centers for Medicare & Medicaid Services.
C. Nationally Non-Covered Indications
All other indications for the use of VADs not otherwise listed remain non-covered, except in the context of Category B investigational device exemption clinical trials (42 CFR 405) or as a routine cost in clinical trials defined under section 310.1 of the National Coverage Determinations (NCD) Manual.
D. Other
This policy does not address coverage of VADs for right ventricular support, biventricular support, use in beneficiaries under the age of 18, use in beneficiaries with complex congenital heart disease, or use in beneficiaries with acute heart failure without a history of chronic heart failure. Coverage under section 1862(a)(1)(A) of the Act for VADs in these situations will be made by local Medicare Administrative Contractors within their respective jurisdictions.


